Adaptation is arguably the most critical biological process in the evolution of species. The process of evolution by natural selection is the cornerstone of evolutionary biology (and indeed, all of contemporary biology!) and adaptation remains fundamental to the process. We know that adaptation is based on the idea that some genetic variants are ‘better’ adapted than others, and thus are unequally shared across a population. But where does this genetic variation come from?
The accumulation of new genetic variation
The classic way for new genetic variants to appear is often thought of as mutation: changes in a single base in the DNA are caused by various external processes such as chemical, physical or environmental influences (such as the sci-fi classics like UV rays or toxic chemicals). Although these forms of mutations happen very rarely and certainly don’t have the same effects comic books would leave you to believe, new mutations can occur relatively rapidly depending on the characteristics of the species. However, the most common way for new mutations to occur is actually part of the DNA replication process: copying DNA is not always perfect and even though the relevant proteins essentially run a spellcheck, sometimes the copy is not 100% perfect and new mutations occur.

It is important to remember that only mutations that are present in the reproductive cells (sperm and eggs) can be inherited and passed on, and thus be a source for adaptation. Mutations in other tissues of the body, such as within the skin, are not spread across the entire body of the subject and thus aren’t passed on to offspring.
Standing genetic variation
Alternatively, genetic variation might already be present within a species or population. This is more likely if population sizes are large and populations are well connected and interbreeding. We refer to this diverse initial gene pool as ‘standing genetic variation’: that is, the amount of genetic variation within the population or species before the selective pressure requiring adaptation. Standing genetic variation can be thought of as the ‘diversity of choices’ for natural selection to act upon: the variants are readily available, and if a good choice exists it will be favoured by natural selection and become more widespread within the population or species (i.e. evolve).


We’ve discussed standing genetic variation before on The G-CAT, but often in a different light (and phrasing). For example, when we’ve talked about founder effect: that is, when a population is formed from only a few different individuals which causes it to be very genetically depauperate. In populations under strong founder effect, there is very little standing genetic variation for natural selection to act upon. This has long been an enigma for many pest species: how have they managed to proliferate so widely when they often originate from so few individuals and lack genetic diversity?
Adaptive variation
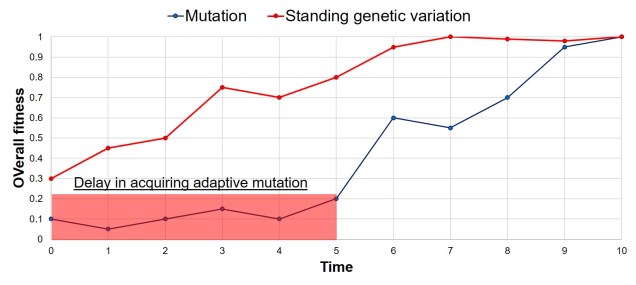
Adaptation may not require new genetic variants to be generated from mutation. If there are a large number of alleles within the gene pool to start with, then natural selection may favour one of those variants over others and allow adaptation to start immediately. Compared to the rate at which new mutations occur, are potentially corrected for in DNA repair, are potentially erased by genetic drift, and then put under selective pressure, adaptation from standing genetic variation can occur very quickly.
Conserving genetic variation
Given the adaptive potential provided by maintaining a good amount of standing genetic variation, it is imperative to conserve genetic diversity within populations in conservation efforts. This is why we often equate genetic diversity with ‘adaptive potential’ of species, although the exact amount of genetic diversity required for adaptive potential depends on a large number of other factors. Clearly, in some instances species show the ability to adapt to new pressures or novel environments even without a large amount of standing genetic variation.
It is important to remember that standing genetic variation consists of two types: neutral genetic diversity, which is not necessarily under selection at the time, and adaptive genetic diversity, which is directly under selection (although this can be either for or against the given variant). However, currently neutral genetic variants may become adaptive variants in the future if selective pressures change: although those different variants aren’t necessarily beneficial or detrimental at the moment, that may change in the future. Thus, conserving both types of genetic diversity is important for the survivability and longevity of populations under conservation programs.
Other types of adaptation
Although genetic diversity is clearly critically important for adaptive potential, alternative mechanisms for adaptation also exist. One of these relies less on the actual genetic variants being different, but rather how individual genes are used. This can happen in a few different ways, but mostly commonly this is through alternative splicing: when a gene is being ‘read’ and a protein is produced, different parts of the gene can be used (and in different order) to make a completely different protein.

Believe it or not, we’ve sort of discussed the effects of alternative splicing before. Phenotypic plasticity occurs when a single organism can have very different physiological traits depending on the environment: even though the genes are the same, they are utilised in different ways to make a different body shape. This is how some species can look incredibly different when they live in different places even if they’re genetically very similar. That said, for the vast majority of species maintaining good levels of genetic diversity is critical for the survivability of said species.