Species which exist in fragmented, isolated and reduced populations have elevated extinction risk. Not only are they more susceptible to demographic and environmental stochasticity, which can easily wipe out small populations, but they also suffer from a range of genetic impacts. Notably, populations often lose significant amounts of genetic diversity as they reduce in size, potentially losing important adaptive diversity enabling them to respond to current and future environmental change. At the same time, random genetic drift becomes stronger relative to natural selection, reducing the efficacy of selection to be able to increase the frequency of favourable alleles and reduce the frequency of maladaptive ones. Together, these impacts create feedback loops which hasten the decline into the extinction vortex.
It should come as no surprise to any reader of The G-CAT that I’m a firm believer against the false dichotomy (and yes, I really do love that phrase) of “nature versus nurture.” Primarily, this is because the phrase gives the impression of some kind of counteracting balance between intrinsic (i.e. usually genetic) and extrinsic (i.e. usually environmental) factors and how they play a role in behaviour, ecology and evolution. While both are undoubtedly critical for adaptation by natural selection, posing this as a black-and-white split removes the possibility of interactive traits.
The real Circle of Life. Not only do genes and the environment interact with one another, but genes may interact with other genes and environments may be complex and multi-faceted.
A very simplified example of adaptation from genetic variation. In this example, we have two different alleles of a single gene (orange and blue). Natural selection favours the blue allele so over time it increases in frequency. The difference between these two alleles is at least one base pair of DNA sequence; this often arises by mutation processes.
Despite how important the underlying genes are for the formation of proteins and definition of physiology, they are not omnipotent in that regard. In fact, many other factors can influence how genetic traits relate to phenotypic traits: we’ve discussed a number of these in minor detail previously. An example includes interactions across different genes: these can be due to physiological traits encoded by the cumulative presence and nature of many loci (as in quantitative trait loci and polygenic adaptation). Alternatively, one gene may translate to multiple different physiological characters if it shows pleiotropy.
Differential expression
One non-direct way genetic information can impact on the phenotype of an organism is through something we’ve briefly discussed before known as differential expression. This is based on the notion that different environmental pressures may affect the expression (that is, how a gene is translated into a protein) in alternative ways. This is a fundamental underpinning of what we call phenotypic plasticity: the concept that despite having the exact same (or very similar) genes and alleles, two clonal individuals can vary in different traits. The is related to the example of genetically-identical twins which are not necessarily physically identical; this could be due to environmental constraints on growth, behaviour or personality.
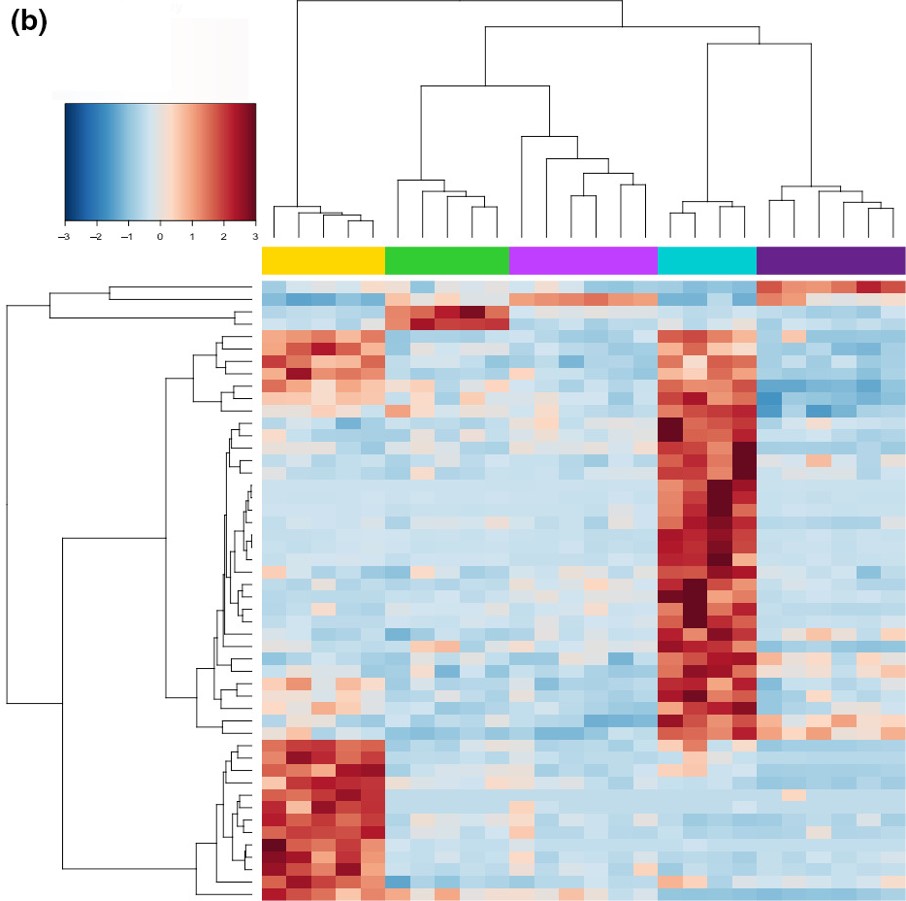
An example of differential expression in wild populations of southern pygmy perch, courtesy of Brauer et al. (2017). In this figure, each column represents a single individual fish, with the phylogenetic tree and coloured boxes at the top indicating the different populations. Each row represents a different gene (this is a subset of 50 from a much larger dataset). The colour of each cell indicates whether the expression of that gene is expressed more (red) or less (blue) than average. As you can see, the different populations can clearly be seen within their expression profiles, with certain genes expressing more or less in certain populations.
The discovery of epigenetic markers and their influence on gene expression has opened up the possibility of understanding heritable traits which don’t appear to be clearly determined by genetics alone. For example, research into epigenetics suggest that heritable major depressive disorder (MDD) may be controlled by the expression of genes, rather than from specific alleles or genetic variants themselves. This is likely true for a number of traits for which the association to genotype is not entirely clear.
Epigenetic adaptation?
From an evolutionary standpoint again, epigenetics can similarly influence the ‘bang for a buck’ of particular genes. Being able to translate a single gene into many different forms, and for this to be linked to environmental conditions, allows organisms to adapt to a variety of new circumstances without the need for specific adaptive genes to be available. Following this logic, epigenetic variation might be critically important for species with naturally (or unnaturally) low genetic diversity to adapt into the future and survive in an ever-changing world. Thus, epigenetic information might paint a more optimistic outlook for the future: although genetic variation is, without a doubt, one of the most fundamental aspects of adaptability, even horrendously genetically depleted populations and species might still be able to be saved with the right epigenetic diversity.
A relatively simplified example of adaptation from epigenetic variation. In this example, we have a species of cat; the ‘default’ cat has non-tufted ears and an orange coat. These two traits are controlled by the expression of Genes A and B, respectively: in the top cat, neither gene is expressed. However, when this cat is placed into different environments, the different genes are “switched on” by epigenetic factors (the green markers). In a rainforest environment, the dark foliage makes darker coat colour more adaptive; switching on Gene B allows this to happen. Conversely, in a desert environment switching on Gene A causes the cat to develop tufts on its ears, which makes it more effective at hunting prey hiding in the sands. Note that in both circumstances, the underlying genetic sequence (indicated by the colours in the DNA) is identical: only the expression of those genes change.
Epigenetic research, especially from an ecological/evolutionary perspective, is a very new field. Our understanding of how epigenetic factors translate into adaptability, the relative performance of epigenetic vs. genetic diversity in driving adaptability, and how limited heritability plays a role in adaptation is currently limited. As with many avenues of research, further studies in different contexts, experiments and scopes will reveal further this exciting new aspect of evolutionary and conservation genetics. In short: watch this space! And remember, ‘nature is nurture’ (and vice versa)!
Managing and conserving threatened and endangered species in the wild is a difficult process. There are a large number of possible threats, outcomes, and it’s often not clear which of these (or how many of these) are at play at any one given time. Thankfully, there are also a large number of possible conservation tools that we might be able to use to protect, bolster and restore species at risk.
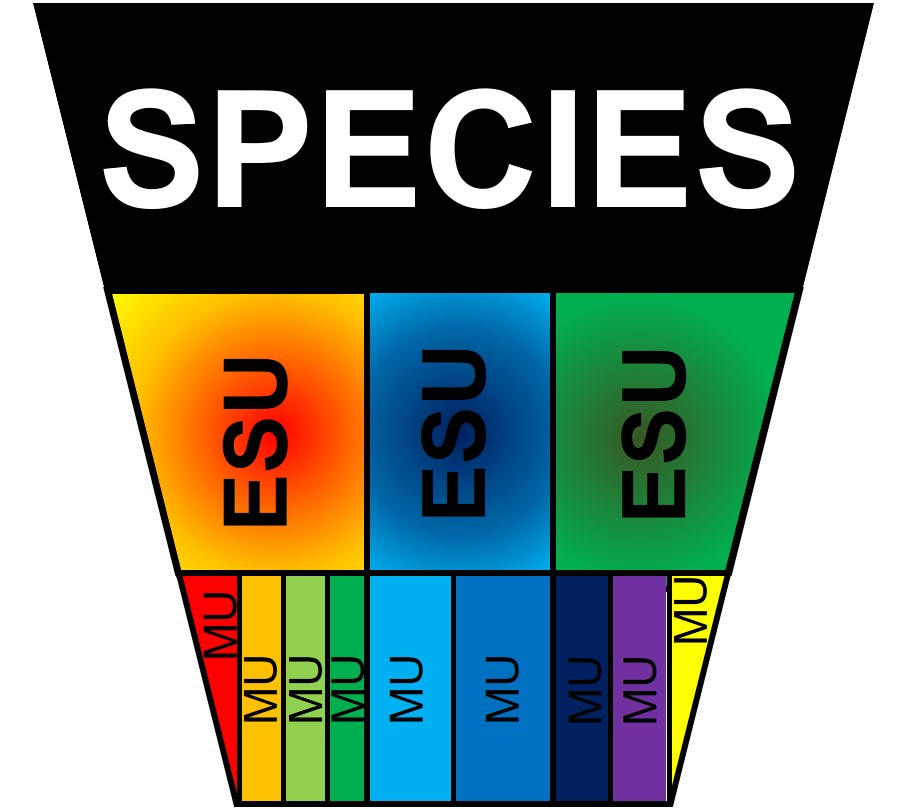
A diagram of the hierarchy of structure within a species. Remember that ESUs, by definition, should be evolutionary different from one another (i.e. adaptively divergent) whilst MUs are not necessarily divergent to the same degree.
This can lead to a particular paradigm of conservation management: keeping everything separate and pure is ‘best practice’. The logic is that, as these different groups have evolved slightly differently from one another (although there is often a lot of grey area about ‘differently enough’), mixing these groups together is a bad idea. Particularly, this is relevant when we consider translocations (“it’s never acceptable to move an organism from one ESU into another”) and captive breeding programs (“it’s never acceptable to breed two organisms together from different ESUs”). So, why not? Why does it matter if they’re a little different?
Outbreeding depression
Well, the classic reasoning is based on a concept called ‘outbreeding depression’. We’ve mentioned outbreeding depression before, and it is a key concept kept in mind when developing conservation programs. The simplest explanation for outbreeding depression is that evolution, through the strict process of natural selection, has pushed particularly populations to evolve certain genetic variants for a certain selective pressure. These can vary across populations, and it may mean that populations are locally adapted to a specific set of environmental conditions, with the specific set of genetic variants that best allow them to do this.
However, when you mix in the genetic variants that have evolved in a different population, by introducing a foreign individual and allowing them to breed, you essentially ‘tarnish’ the ‘pure’ gene pool of that population with what could be very bad (maladaptive) genes. The hybrid offspring of ‘native’ and this foreign individual will be less adaptive than their ‘pure native’ counterparts, and the overall adaptiveness of the population will decrease as those new variants spread (depending on the number introduced, and how negative those variants are).
An example of how outbreeding depression can affect a species. The original red fish population is not doing well- it is of conservation concern, and has very little genetic diversity (only the blue gene in this example). So, we decide to introduce new genetic diversity by adding in green fish, which have the orange gene. However, the mixture of the two genes and the maladaptive nature of the orange gene actually makes the situation worse, with the offspring showing less fitness than their preceding generations.
You might be familiar with inbreeding depression, which is based on the loss of genetic diversity from having too similar individuals breeding together to produce very genetically ‘weak’ offspring through inbreeding. Outbreeding depression could be thought of as the opposite extreme; breeding too different individuals introduced too many ‘bad’ alleles into the population, diluting the ‘good’ alleles.
An overly simplistic representation of how inbreeding and outbreeding depression can reduce overall fitness of a species. In inbreeding depression, the lack of genetic diversity due to related individuals breeding with one another makes them at risk of being unable to adapt to new pressures. Contrastingly, adding in new genes from external populations which aren’t fit for the target population can also reduce overall fitness by ‘diluting’ natural, adaptive allele frequencies in the population.
Genetic rescue
It might sound awfully purist to only preserve the local genetic diversity, and to assume that any new variants could be bad and tarnish the gene pool. And, surprisingly enough, this is an area of great debate within conservation genetics.
An example of genetic rescue. This circumstance is identical to the one above, with the key difference being in the fitness of the introduced gene. The orange gene in this example is actually beneficial to the target population: by providing a new, adaptive allele for natural selection to act upon, overall fitness is increased for the red fish population.
The balance
So, what’s the balance between the two? Is introducing new genetic variation a bad idea, and going to lead to outbreeding depression; or a good idea, and lead to genetic rescue? Of course, many of the details surrounding the translocation of new genetic material is important: how different are the populations? How different are the environments (i.e. natural selection) between them? How well will the target population take up new individuals and genes?
Overall, however, the more recent and well-supported conclusion is that fears regarding outbreeding depression are often strongly exaggerated. Bad alleles that have been introduced into a population can be rapidlypurged by natural selection, and the likelihood of a strongly maladaptive allele spreading throughout the population is unlikely. Secondly, given the lack of genetic diversity in the target population, most that need the genetic rescue are so badly maladaptive as it is (due to genetic drift and lack of available adaptive alleles) that introducing new variants is unlikely to make the situation much worse.
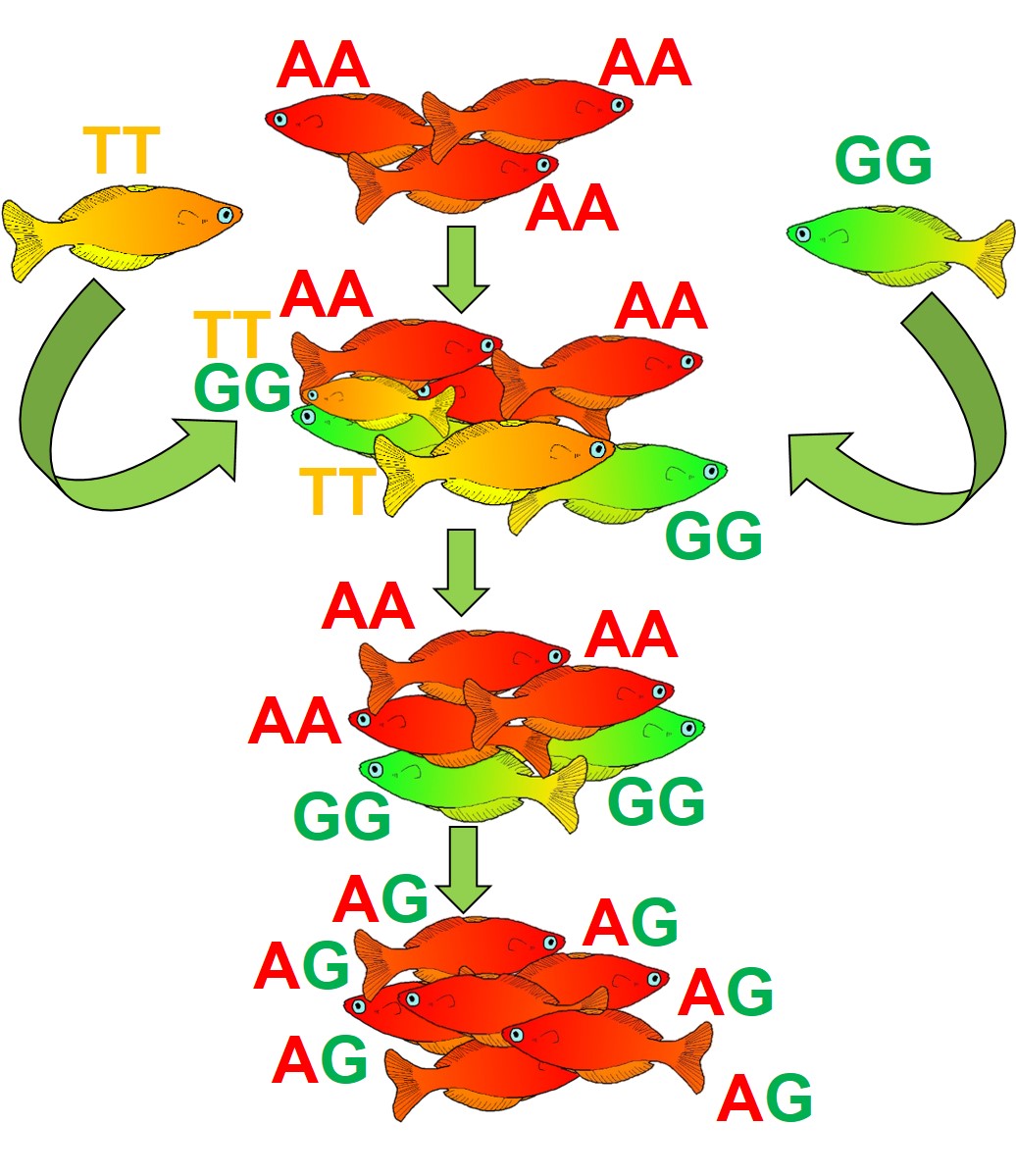
An example of how introducing maladaptive alleles might not necessarily lead to decreased fitness. In this example, we again start with our low diversity red fish population, with only one allele (AA). To help boost genetic diversity, we introduce orange fish (with the TT allele) and green fish (with the GG allele) into the population. However, the TT allele is not very adaptive in this new environment, and individuals with the TT gene quickly die out (i.e. be ‘purged’). Individual with the GG gene, however, do well, and continue to integrate into the red population. Over time, these two variants will mix together as the two populations hybridise and overall fitness will increase for the population.
That said, outbreeding depression is not an entirely trivial concept and there are always limitations in genetic rescue procedures. For example, it would be considered a bad idea to mix two different species together and make hybrids, since the difference between two species, compared to two populations, can be a lot stronger and not necessarily a very ‘natural’ process (whereas populations can mix and disjoin relatively regularly).
The reality of conservation management
Conservation science is, at its core, a crisis discipline. It exists solely as an emergency response to the rapid extinction of species and loss of biodiversity across the globe. The time spent trying to evaluate the risk of outbreeding depression – instead of immediately developing genetic rescue programs – can cause species to tick over to the afterlife before we get a clear answer. Although careful consideration and analysis is a requirement of any good conservation program, preventing action due to almost paranoid fear is not a luxury endangered species can afford.
Given the strong influence of genetic identity on the process and outcomes of the speciation process, it seems a natural connection to use genetic information to study speciation and species identities. There is a plethora of genetics-based tools we can use to investigate how speciation occurs (both the evolutionary processes and the external influences that drive it). One clear way to test whether two populations of a particular species are actually two different species is to investigate genes related to reproductive isolation: if the genetic differences demonstrate reproductive incompatibilities across the two populations, then there is strong evidence that they are separate species (at least under the Biological Species Concept; see Part One for why!). But this type of analysis requires several tools: 1) knowledge of the specific genes related to reproduction (e.g. formation of sperm and eggs, genital morphology, etc.), 2) the complete and annotated genome of the species (to be able to find and analyse the right genes properly) and 3) a good amount of data for the populations in question. As you can imagine, for people working on non-model species (i.e. ones that haven’t had the same history and detail of research as, say, humans and mice), this can be problematic. So, instead, we can use other genetic information to investigate and suggest patterns and processes related to the formation of new species.
Is reproductive isolation naturally selected for or just a consequence?
A fundamental aspect of studies of speciation is a “chicken or the egg”-type paradigm: does natural selection directly select for rapid reproductive isolation, preventing interbreeding; or as a secondary consequence of general adaptive differences, over a long history of evolution? This might be a confusing distinction, so we’ll dive into it a little more.
Of the two proposed models of speciation, the by-product of natural selection (the second model) has been the more favoured. Simply put, this expands on Darwin’s theory of evolution that describes two populations of a single species evolving independently of one another. As these become more and more different, both in physical (‘phenotype’) and genetic (‘genotype’) characteristics, there comes a turning point where they are so different that an individual from one population could not reasonably breed with an individual from the other to form a fertile offspring. This could be due to genetic incompatibilities (such as different chromosome numbers), physiological differences (such as changes in genital morphology), or behavioural conflicts (such as solitary vs. group living).
Certainly, this process makes sense, although it is debatable how fast reproductive isolation would occur in a given species (or whether it is predictable just based on the level of differentiation between two populations). Another model suggests that reproductive isolation actually might arise very quickly if natural selection favours maintaining particular combinations of traits together. This can happen if hybrids between two populations are not particularly well adapted (fit), causing natural selection to favour populations to breed within each group rather than across groups (leading to reproductive isolation). Typically, this is referred to as ‘reinforcement’ and predominantly involves isolating mechanisms that prevent individuals across populations from breeding in the first place (since this would be wasted energy and resources producing unfit offspring). The main difference between these two models is the sequence of events: do populations ecologically diverge, and because of that then become reproductively isolated, or do populations selectively breed (enforcing reproductive isolation) and thus then evolve independently?
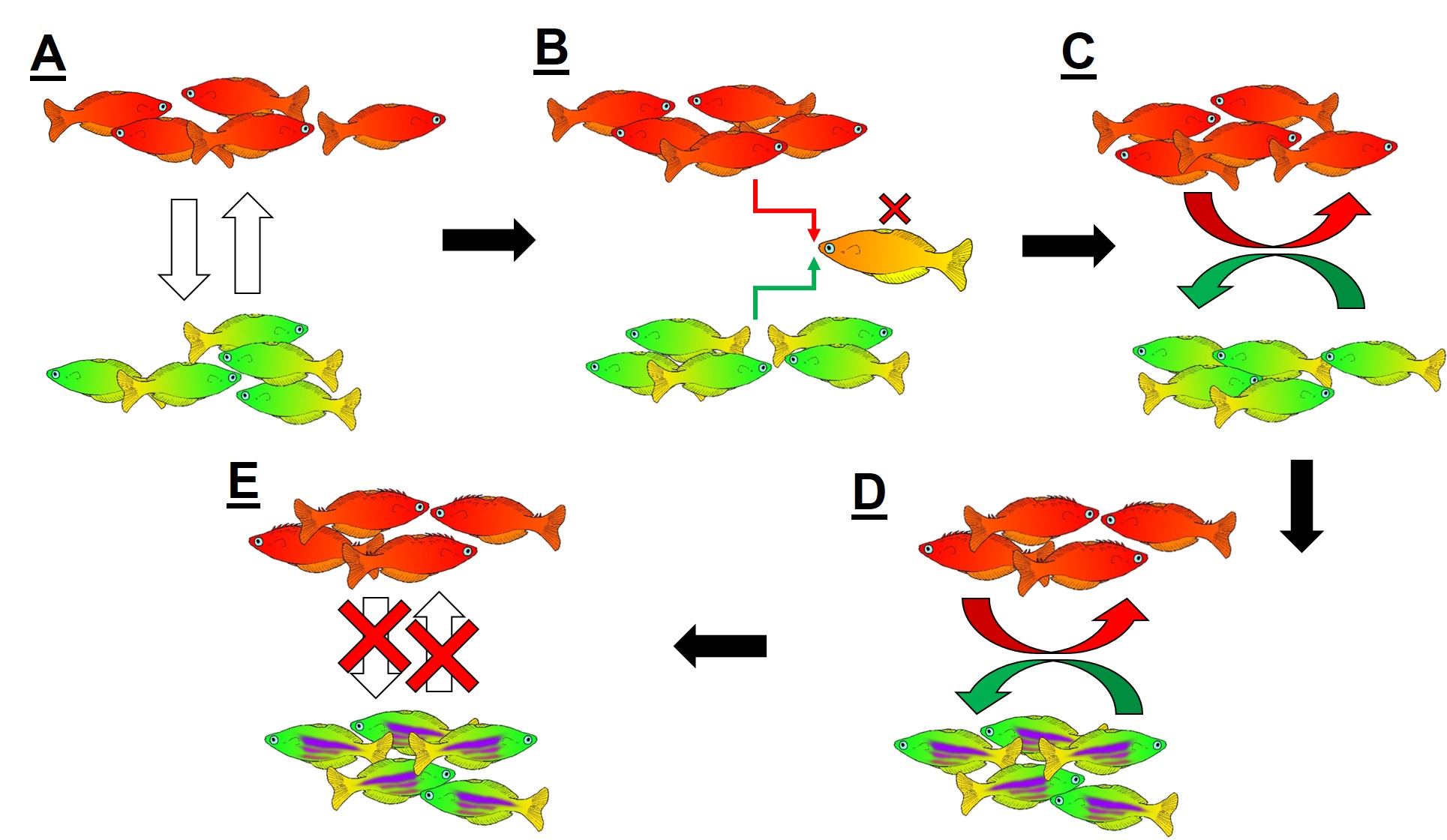
An example of reinforcement leading to speciation. A) We start with two populations of a single species (a red fish population and a green fish population), which can interbreed (the arrows). B) Because these two groups can breed, hybrids of the two populations can be formed. However, due to the poor combination of red and green fish genes within a hybrid, they are not overly fit (the red cross). C) Since natural selection doesn’t favour forming hybrids, populations then adapt to selectively breed only with similar fish, reducing the amount of interbreeding that occurs. D) With the two populations effectively isolated from one another, different adaptations specific to each population (spines in red fish, purple stripes in green fish) can evolve, causing them to further differentiate. E) At some point in the differentiation process, hybrids move from being just selectively unfit (as in B)) to entirely impossible, thus making the two populations formal species. In this example, evolution has directly selected against hybrids first, thus then allowing ecological differences to occur (as opposed to the other way around).
Reproductive isolation through DMIs
The reproductive incompatibility of two populations (thus making them species) is often intrinsically linked to the genetic make-up of those two species. Some conflicts in the genetics of Population 1 and Population 2 may mean that a hybrid having half Population 1 genes and half Population 2 genes will have serious fitness problems (such as sterility or developmental problems). Dramatic genetic differences, particularly a difference in the number of chromosomes between the two sources, is a significant component of reproductive isolation and is usually to blame for sterile hybrids such as ligers, zorse and mules.
However, subtler genetic differences can also have a strong effect: for example, the unique combination of Population 1 and Population 2 genes within a hybrid might interact with one another negatively and cause serious detrimental effects. These are referred to as “Dobzhansky-Müller Incompatibilities” (DMIs) and are expected to accumulate as the two populations become more genetically differentiated from one another. This can be a little complicated to imagine (and is based upon mathematical models), but the basis of the concept is that some combinations of gene variants have never, over evolutionary history, been tested together as the two populations diverge. Hybridisation of these two populations suddenly makes brand new combinations of genes, some of which may be have profound physiological impacts (including on reproduction).
An example of how Dobzhansky-Müller Incompatibilities arise, adapted from Coyne & Orr (2004). We start with an initial population (center top), which splits into two separate populations. In this example, we’ll look at how 5 genes (each letter = one gene) change over time in the separate populations, with the original allele of the gene (lowercase) occasionally mutating into a new allele (upper case). These mutations happen at random times and in random genes in each population (the red letters), such that the two become very different over time. After a while, these two populations might form hybrids; however, given the number of changes in each population, this hybrid might have some combinations of alleles that are ‘untested’ in their evolutionary history (see below). These untested combinations may cause the hybrid to be infertile or unviable, making the two populations isolated species.
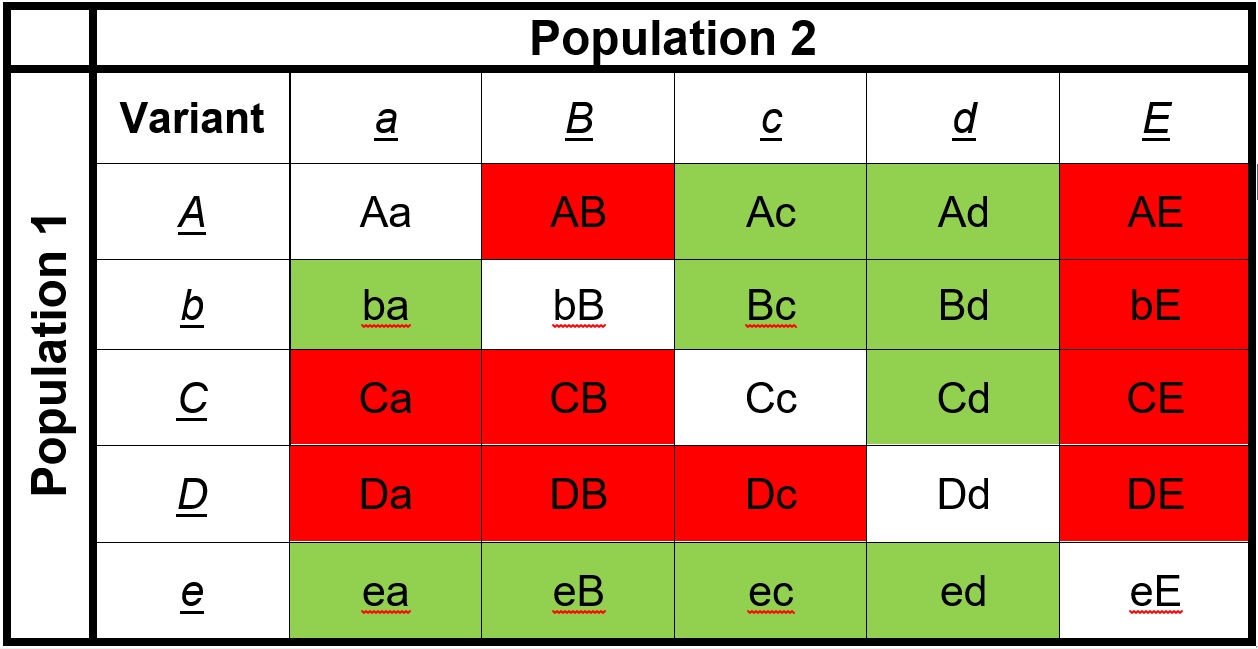
The list of ‘untested’ genetic combinations from the above example. This table shows the different combinations of each gene that could be made in a hybrid if these two populations interbred. The red cells indicate combinations that have never been ‘tested’ together; that is, at no point in the evolutionary history of these two populations were those two particular alleles together in the same individual. Green cells indicate ones that were together at some point, and thus are expected to be viable combinations (since the resultant populations are obviously alive and breeding).
How can we look at speciation in action?
We can study the process of speciation in the natural world without focussing on the ‘reproductive isolation’ element of species identity as well. For many species, we are unlikely to have the detail (such as an annotated genome and known functions of genes related to reproduction) required to study speciation at this level in any case. Instead, we might choose to focus on the different factors that are currently influencing the process of speciation, such as how the environmental, demographic or adaptive contexts of populations plays a role in the formation of new species. Many of these questions fall within the domain of phylogeography; particularly, how the historical environment has shaped the diversity of populations and species today.
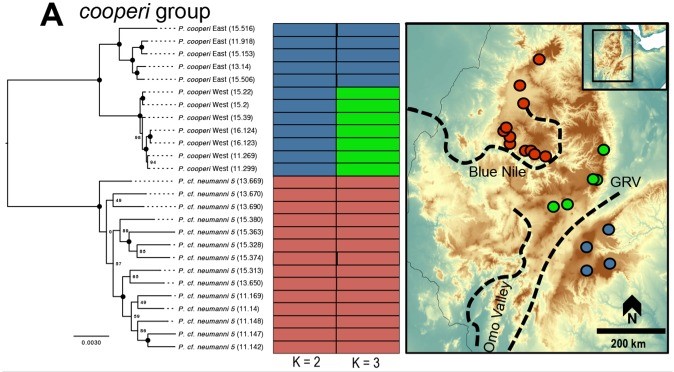
An example of the interplay between speciation and phylogeography, taken from Reyes-Velasco et al. (2018). They investigated the phylogeographic history of several different groups of species within the frog genus Ptychadena; in this figure, we can see how the different species (indicated by the colours and tree on the left) relate to the geography of their habitat (right).
A variety of different analytical techniques can be used to build a picture of the speciation process for closely related or incipient species. A good starting point for any speciation study is to look at how the different study populations are adapting; is there evidence that natural selection is pushing these populations towards different genotypes or ecological niches? If so, then this might be a precursor for speciation, and we can build on this inference with other complementary analyses.
For example, estimating divergence times between populations can help us suggest whether there has been sufficient time for speciation to occur (although this isn’t always clear cut). Additionally, we could estimate the levels of genetic hybridisation (‘introgression’) between two populations to suggest whether they are reasonably isolated and divergent enough to be considered functional species.
The future of speciation genomics
Although these can help answer some questions related to speciation, new tools are constantly needed to provide a clearer picture of the process. Understanding how and why new species are formed is a critical aspect of understanding the world’s biodiversity. How can we predict if a population will speciate at some point? What environmental factors are most important for driving the formation of new species? How stable are species identities, really? These questions (and many more) remain elusive for a wide variety of life on Earth.