The roles of aridification and sea level changes in the diversification and persistence of freshwater fish lineages
The process of publishing science is a lengthy one – there are many rounds of revisions, assessments, and review required before a paper can be published. With that, I’m very proud to announce that the first paper from my PhD has recently been published in the journal Molecular Ecology. This paper is a collection of a lot of complex analyses, and addressing some relatively complicated biogeographical questions, so I’ve decided to provide a simplified summary here.
We’ve spent some time before discussing the nature of the term ‘species’ and what it means in reality. Of course, answers to questions in biology are always more complicated than we wish they might be, and despite the common nomenclature of the word ‘species’ the underlying definition is convoluted and variable.
For anyone who has had to study geography at some point in their education, you’d likely be familiar with the idea of river courses drawn on a map. They’re so important, in fact, that they are often the delimiting factor in the edges of countries, states or other political units. Water is a fundamental requirement of all forms of life and the riverways that scatter the globe underpin the maintenance, structure and accumulation of a large swathe of biodiversity.
To expand on this, we’re going to look at a few different models of how the spatial distribution of populations influences their divergence, and particularly how these factor into different processes of speciation.
What comes first, ecological or genetic divergence?
The order of these two processes have been in debate for some time, and different aspects of species and the environment can influence how (or if) these processes occur.
Different spatial models of speciation
Generally, when we consider the spatial models for speciation we divide these into distinct categories based on the physical distance of populations from one another. Although there is naturally a lot of grey area (as there is with almost everything in biological science), these broad concepts help us to define and determine how speciation is occurring in the wild.
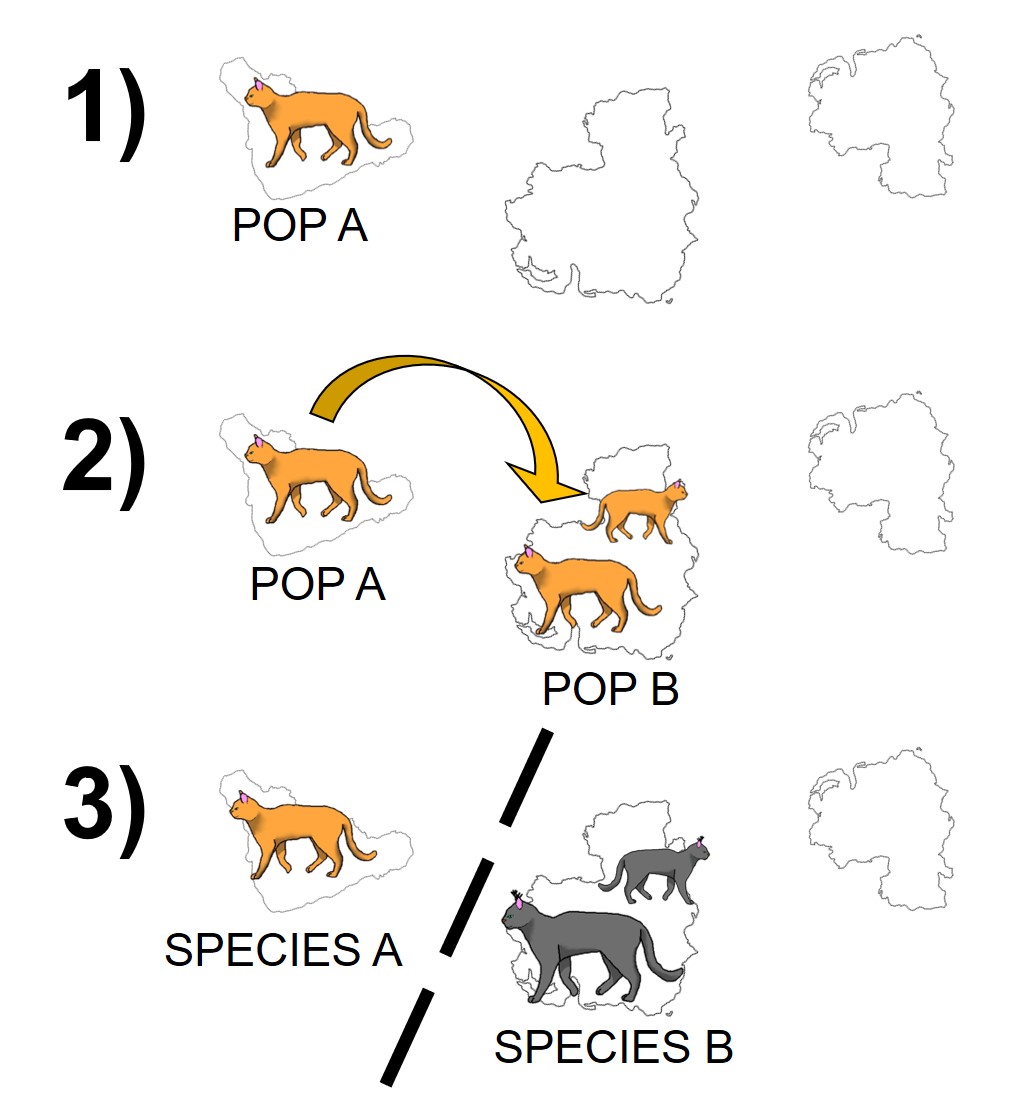
The standard model of allopatric speciation, following an island model. 1) We start with a single population occupying a single island. 2) A rare dispersal event pushes some individuals onto a new island, forming a second population. Note that this doesn’t happen often enough to allow for consistent gene flow (i.e. the island was only colonised once). 3) Over time, these populations may accumulate independent genetic and ecological changes due to both natural selection and drift, and when they become so different that they are reproductively isolated they can be considered separate species.
A step closer in bringing populations geographically together in speciation is “parapatry” and “peripatry”. Parapatric populations are often geographically close together but not overlapping: generally, the edges of their distributions are touching but do not overlap one another. A good analogy would be to think of countries that share a common border. Parapatry can occur when a species is distributed across a broad area, but some form of narrow barrier cleaves the distribution in two: this can be the case across particular environmental gradients where two extremes are preferred over the middle.
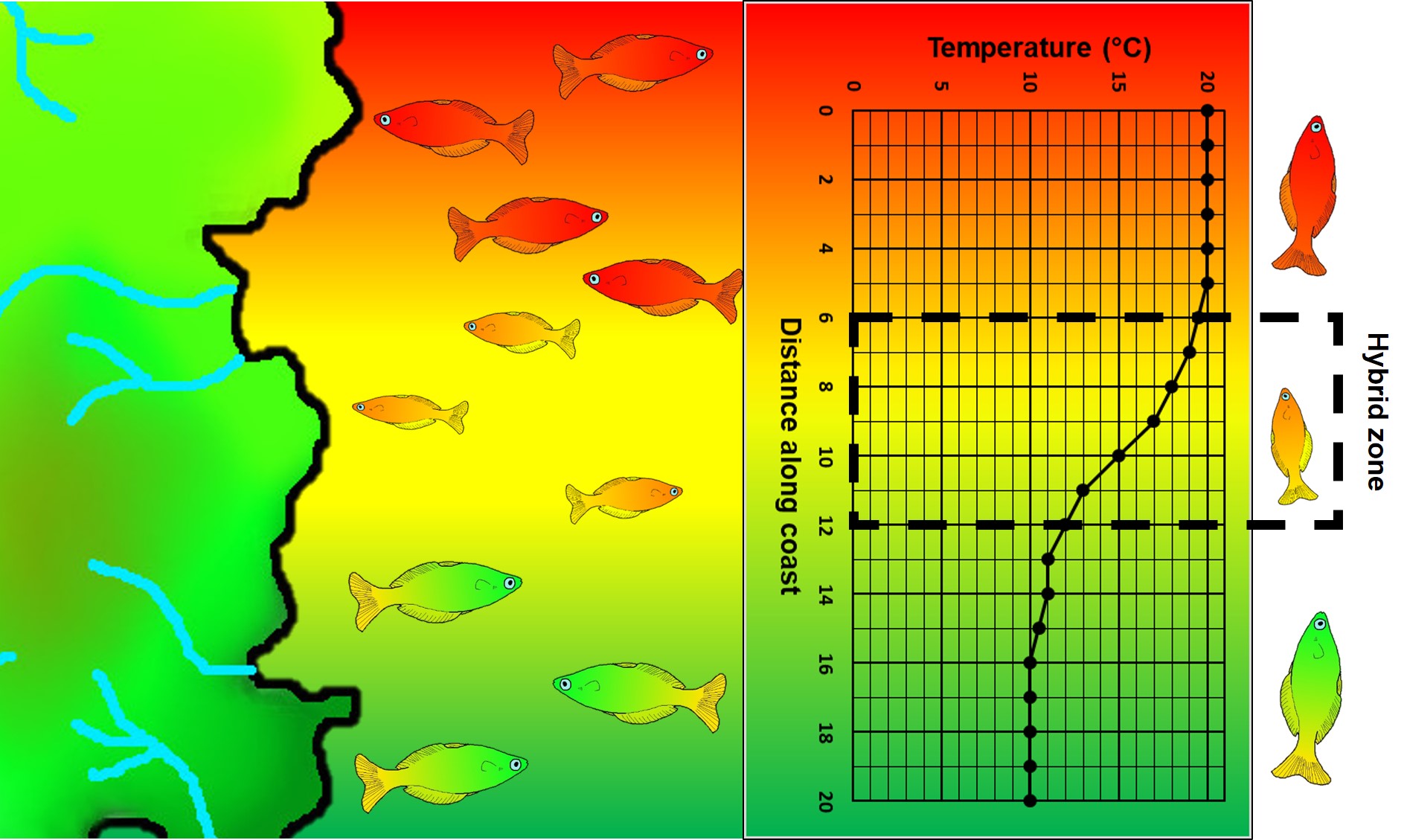
An example of parapatric species across an environment gradient (in this case, a temperature gradient along the ocean coastline). Left: We have two main species (red and green fish) which are adapted to either hotter or colder temperatures (red and green in the gradient), respectively. A small zone of overlap exists where hybrid fish (yellow) occur due to intermediate temperature. Right: How the temperature varies across the system, forming a steep gradient between hot and cold waters.
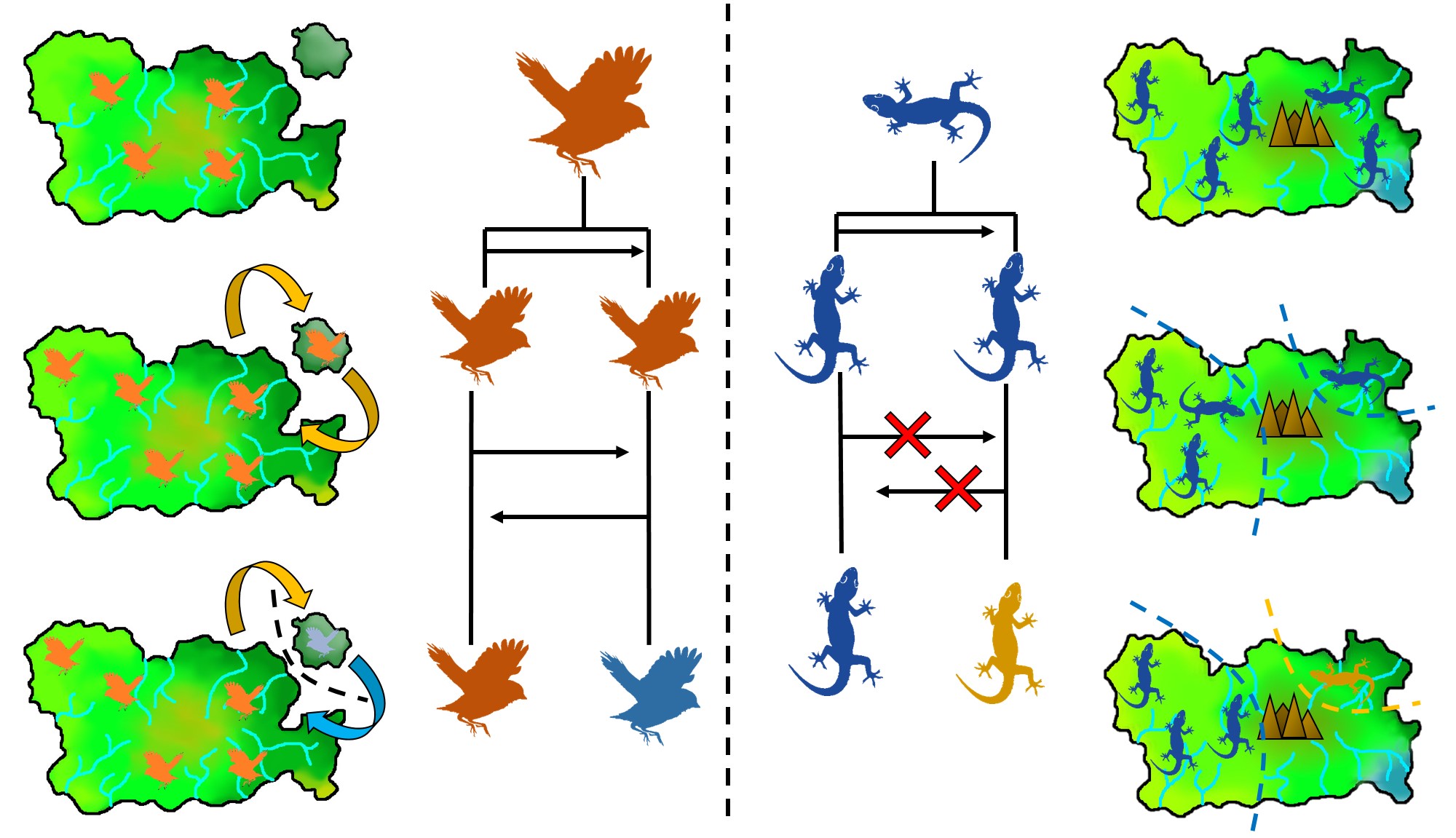
The two main ways peripatric species can form. Left: The dispersal method. In this example, there is a central ‘source’ population (orange birds on the main island), which holds most of the distribution. However, occasionally (more frequently than in the allopatric example above) birds can disperse over to the smaller island, forming a (mostly) independent secondary population. If the gene flow between this population and the central population doesn’t overwhelm the divergence between the two populations (due to selection and drift), then a new species (blue birds) can form despite the gene flow. Right: The range contraction method. In this example, we start with a single widespread population (blue lizards) which has a rapid reduction in its range. However, during this contraction one population is separated from the main body (i.e. as a refugia), which may also be a precursor of peripatric speciation.
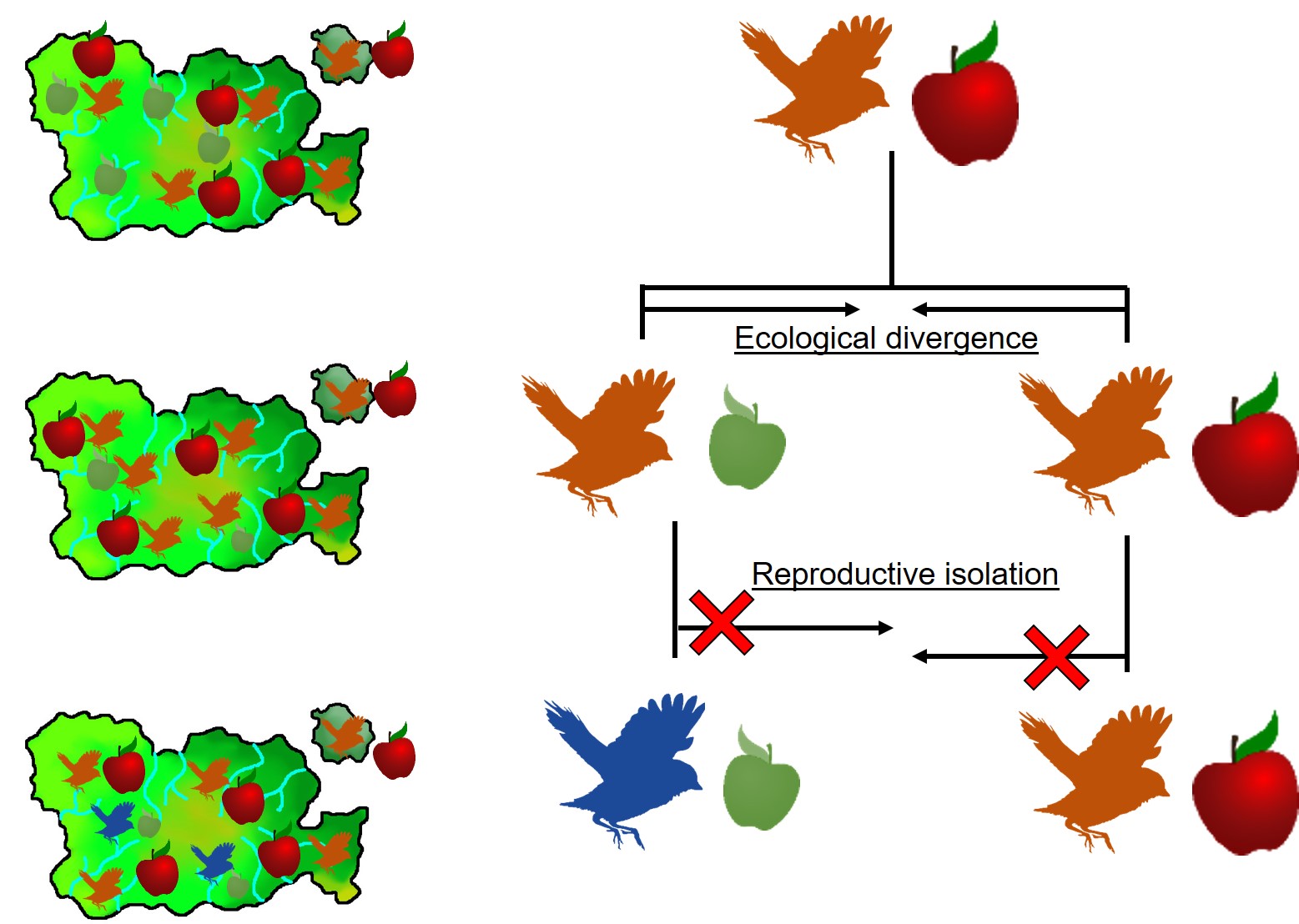
This can be tricky to visualise, so let’s invent an example. Say we have a tropical island, which is occupied by one bird species. This bird prefers to eat the large native fruit of the island, although there is another fruit tree which produces smaller fruits. However, there’s only so much space and eventually there are too many birds for the number of large fruit trees available. So, some birds are pushed to eat the smaller fruit, and adapt to a different diet, changing physiology over time to better acquire their new food and obtain nutrients. This shift in ecological niche causes the two populations to become genetically separated as small-fruit-eating-birds interact more with other small-fruit-eating-birds than large-fruit-eating-birds. Over time, these divergences in genetics and ecology causes the two populations to form reproductively isolated species despite occupying the same island.
A diagram of the ecological speciation example given above. Note that ecological divergence occurs first, with some birds of the original species shifting to the new food source (‘ecological niche’) which then leads to speciation. An important requirement for this is that gene flow is somehow (even if not totally) impeded by the ecological divergence: this could be due to birds preferring to mate exclusively with other birds that share the same food type; different breeding seasons associated with food resources; or other isolating mechanisms.
As you can see, the processes and context driving speciation are complex to unravel and many factors play a role in the transition from population to species. Understanding the factors that drive the formation of new species is critical to understanding not just how evolution works, but also in how new diversity is generated and maintained across the globe (and how that might change in the future).