When I was younger, I used to love visiting our local creek: it was a beautiful spot of nature a short walk from home. On a couple occasions, my Dad took me to the creek to catch yabbies – for a suburban kid, it was one of the few times I actually held and interacted with wild biodiversity, and helped foster my love for conservation and inquiry into biology. In the late 2000s to early 2010s, a likely combination of local pollution and extensive drought extirpated the yabbies from the creek – I would never see one in that creek again. I was devastated for the local loss of a fascinating creature, and the connection to nature it represented, but felt powerless to remedy the situation. To my knowledge, there are still no yabbies in that creek.
It’s been a brief while (oops!) since the last post on The G-CAT (I promise more content is coming soon!), so I thought I would give a quick research update. This week, I had the absolute privilege of attending the 3rd International Conservation Translocation Conference 2023here in sunny Perth (Fremantle, to be more specific). Hosted through the IUCN Species Survival Commission, and particularly the IUCN SSC Conservation Translocation Specialist Group, the conference brought hundreds of attendees from across the globe to share the trials and tribulations of conservation translocation efforts.
Species which exist in fragmented, isolated and reduced populations have elevated extinction risk. Not only are they more susceptible to demographic and environmental stochasticity, which can easily wipe out small populations, but they also suffer from a range of genetic impacts. Notably, populations often lose significant amounts of genetic diversity as they reduce in size, potentially losing important adaptive diversity enabling them to respond to current and future environmental change. At the same time, random genetic drift becomes stronger relative to natural selection, reducing the efficacy of selection to be able to increase the frequency of favourable alleles and reduce the frequency of maladaptive ones. Together, these impacts create feedback loops which hasten the decline into the extinction vortex.
The classic way for new genetic variants to appear is often thought of as mutation: changes in a single base in the DNA are caused by various external processes such as chemical, physical or environmental influences (such as the sci-fi classics like UV rays or toxic chemicals). Although these forms of mutations happen very rarely and certainly don’t have the same effects comic books would leave you to believe, new mutations can occur relatively rapidly depending on the characteristics of the species. However, the most common way for new mutations to occur is actually part of the DNA replication process: copying DNA is not always perfect and even though the relevant proteins essentially run a spellcheck, sometimes the copy is not 100% perfect and new mutations occur.
An example of how adaptation can occur from a new mutation. In this example, we have one gene (TTXTT), with initial only one allele (variant), TTATT. In the second generation (row), a mutation occurs in one individual which creates a new, second allele: TTGTT. This allele is favoured over the TTATT allele, and in the next generation it’s frequency increases as the alternative allele frequency decreases (the pattern is shown in the frequency values on the right side).
Alternatively, genetic variation might already be present within a species or population. This is more likely if population sizes are large and populations are well connected and interbreeding. We refer to this diverse initial gene pool as ‘standing genetic variation’: that is, the amount of genetic variation within the population or species before the selective pressure requiring adaptation. Standing genetic variation can be thought of as the ‘diversity of choices’ for natural selection to act upon: the variants are readily available, and if a good choice exists it will be favoured by natural selection and become more widespread within the population or species (i.e. evolve).
A slightly more complex example of how adaptation can occur from standing variation, this time with two different genes. One codes for fur colour, with two different alleles: GCATA codes for orange fur, and GCGTA codes for grey fur. The other gene codes for ear tufts, with TTCCT coding for tufts and TCCCT coding for no tufts. Natural selection favours both orange fur and tufted ears, and cats with these traits reproduce more frequently than those without (see graph below). These cats probably look familiar.
The frequency of all four alleles (i.e. either allele for both genes) over the generations in the above figure. Clearly, we can see how adaptation rapidly favours orange fur and tufted ears over grey fur and non-tufted ears with the shifts in frequencies over the different alleles.
We’ve discussed standing genetic variation before on The G-CAT, but often in a different light (and phrasing). For example, when we’ve talked about founder effect: that is, when a population is formed from only a few different individuals which causes it to be very genetically depauperate. In populations under strong founder effect, there is very little standing genetic variation for natural selection to act upon. This has long been an enigma for many pest species: how have they managed to proliferate so widely when they often originate from so few individuals and lack genetic diversity?
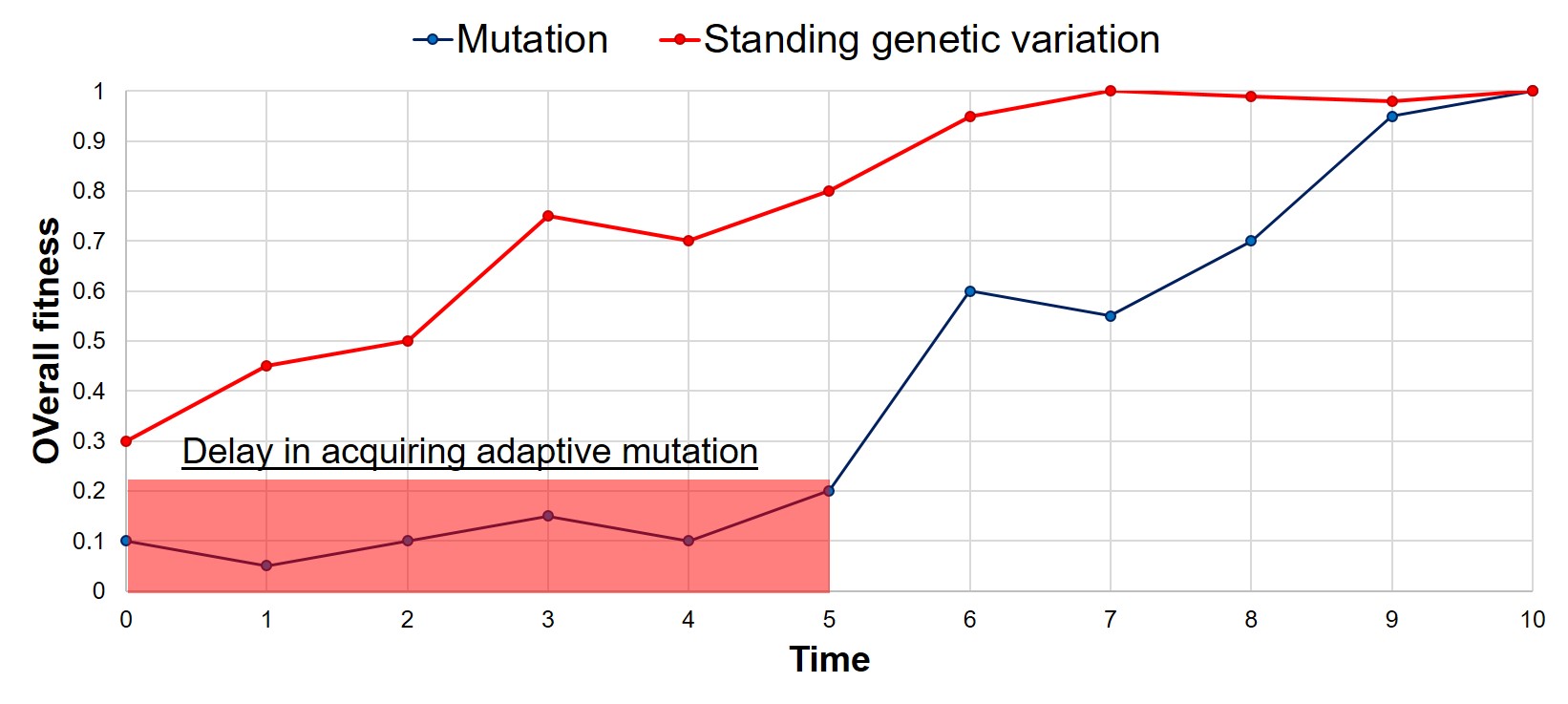
A rough example of the speed of adaptation depending on how the adaptive allele originated: whether it was already present (in the form of standing variation), or whether it was created by a new mutation. As one would expect, there is a significant lag delay in adaptation in the mutation scenario, based on the time it takes for said adaptive mutation to be created through relatively random processes. Thus, a positively selected allele from standing variation can allow a species to adapt much faster than waiting for a positive mutation to occur.
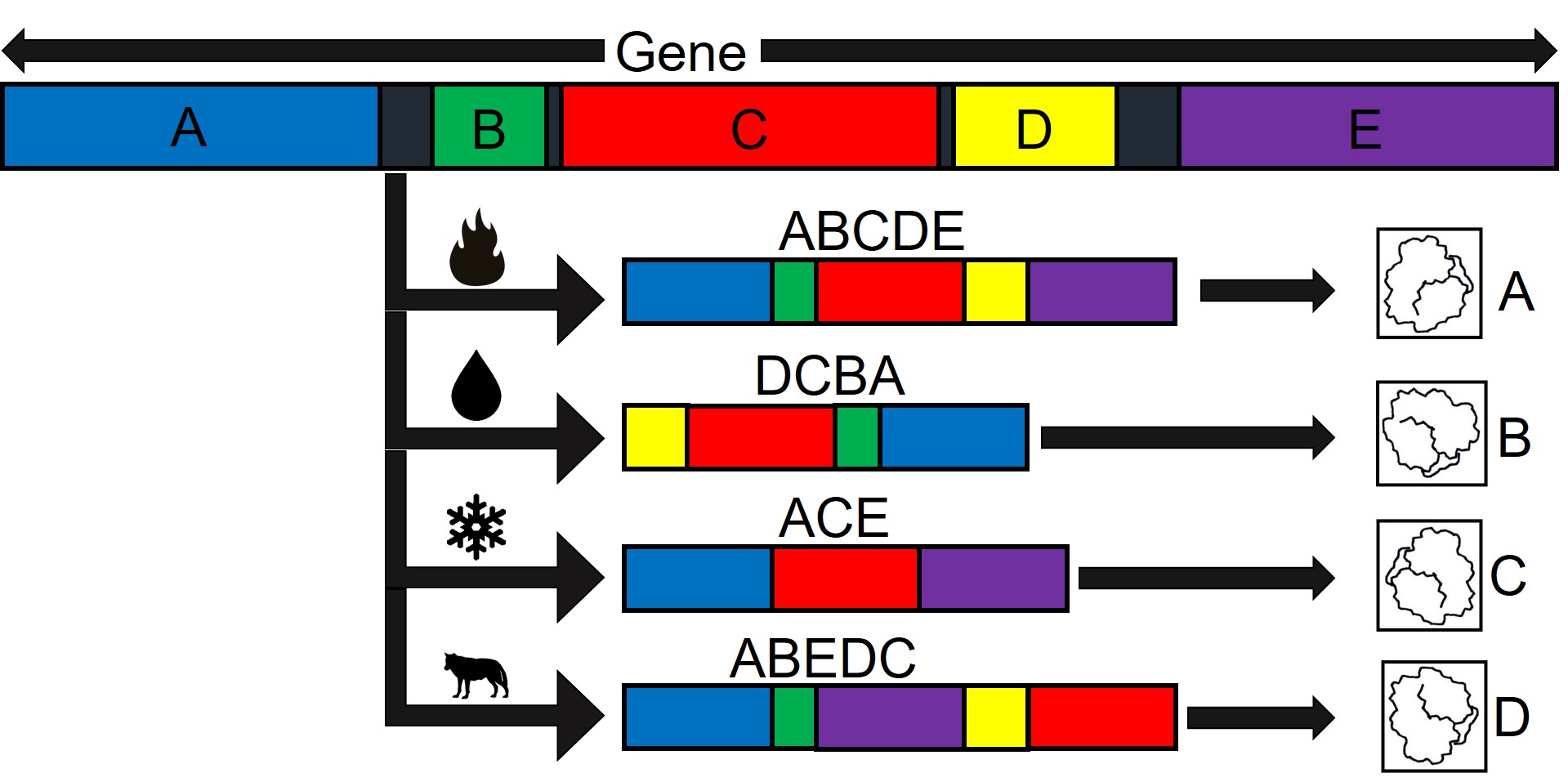
An extreme example of alternate splicing of one gene. We start with a single gene, composed of 5 (A–E) main gene elements (exons). Different environmental pressures (like fire risk, flooding, cold weather or predators, for example) cause the organism to use different combinations of these exons to make different proteins (right side; A–D). Actual alternate splicing is not usually this straight-forward (one gene doesn’t conveniently split into four forms depending on the threat), but the process is generally the same.