Building an entire genome
If bigger is better, then biggest is best. Having the genome of a particular study species fully sequenced allows us to potentially look at all of the genetic variation in the entire gene pool: but how do we sequence the entirety of the genome? And what are the benefits of having a whole genome to refer to?
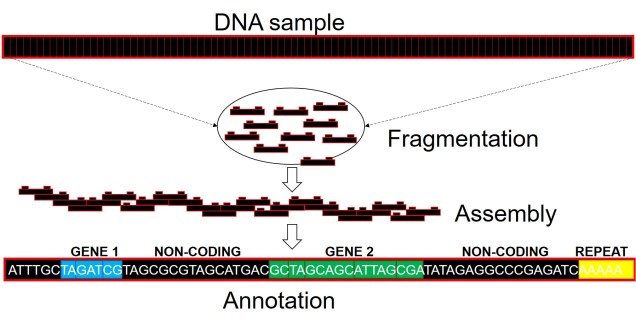
Well, assembling the whole genome of an organism for the first time is a very tricky process. It involves taking DNA sequence from only a few individuals, breaking them down into smaller fragments and multiplying these fragments into the billions (moreorless the same process used in other genomics technologies: the real difference is that we need the full breadth of the genome so that we don’t miss any spaces). From these fragments, we use a complex computer algorithm which builds up a consensus sequence like a Lego tower; by finding parts of sequences which overlap, the software figures out which pieces connect to one another. Hopefully, we eventually end up with one very long continuous sequence; the genome! Sometimes, we might end with a few very large blocks (called contigs), but this is also useful for analyses (correlated with how many/big blocks there are). With this full genome, we use information from other more completed genomes (such as those from model species like humans, mice or even worms) to figure out which sections of the genome relate to specific genes. We can then annotate these sections by labelling them as clear genes, complete with start and end point, and attach a particular physical function of that gene.
The benefits of whole genomes
Having an entire genome as a reference is an extremely helpful tool in conservation and evolutionary studies. The first, and perhaps most obvious benefit, is the sheer scale of the data we can use. By having the entirety of the genome available, we can use potentially billions of base pairs of sequence in our genetic analyses (for reference, the human genome is >3 billion base pairs long). Even if we don’t sequence the full genome for all of our samples, having a reference genome as basis for assembly our reduced datasets significantly improves the quantity and quality of sequences we can use.
Another very important benefit is the ability to prescribe function in our studies. Many of our processes for obtaining data, even for genomic technologies, use random and anonymous fragments of the genome. Although this is a cost-effective way to obtain a very large amount of data, it unfortunately means that we often have no idea which part of the genome our sequences came from. This means that we don’t know which sequences relate to specific genes, and even if we did we would have no idea what those genes are or do! But with an annotated genome, we can take even our fragmented sequence and check it against the genome and find out what genes are present.
Understanding adaptation
Based on that, it seems pretty obvious about exactly how having an annotated genome can help us in studies of adaptation. Knowing the functional aspect of our genetic data allows us to more directly determine how evolution is happening in nature; instead of only being able to say that two species are evolving differently from one another, for example, we can explicitly look at how they are evolving. Is one evolving tolerance to hotter temperatures? Are they evolving different genes to handle different diets? Are they evolving in response to an external influence, like a viral outbreak or changing climate? What are the physiological consequences of these changes? These questions are critical in understanding past and future evolution, and full genome analysis allows us to delve into them much deeper.

This includes allowing us to better understand how adaptation actually works in nature. As we’ve discussed before, more traditional studies often assumed that single, or very few, genes were responsible for allowing a species to adapt and change, and that these genes had very strong effects on their physiology. But what we see far more often is polygenic adaptation; small changes in a very large number of genes which, combined together, allow the species to adapt and evolve. By having the entirety of the genome available, we are much more likely to capture all of the genes that are under natural selection in a particular population or species, painting a clearer picture of their evolutionary trajectory.
Understanding demography
The much larger dataset of full genomes is also important for understanding the non-adaptive parts of evolution; the demographic history. Given that selectively neutral impacts (e.g. reductions in population size) are likely to impact all of the genes in the gene pool somewhat equally, having a full genome allows us to more accurately infer the demographic state and historical patterns of species.
For both adaptive and non-adaptive variation, it is also important to consider what we call linkage disequilibrium. Genetic sequences that are physically close to each other in the genome will often be inherited together due to the imprecision of recombination (a fairly technical process, so I won’t delve into this): what this can mean is that if a gene is under very strong selection, then sequences around this gene will also look like they’re under selection too. This can give falsely positive adaptive genes (i.e. sequences that look like genes under selection but are just linked to a gene that is) or can interfere with demographic analyses (since they often assume no selection, or linkage to selection, on the sequences used). With a whole genome, we can actually estimate how far away a base pair has to be before it’s not linked anymore; we call these linkage blocks, and they’re very useful additions to analyses.

Improving conservation management
In a similar fashion to demography, full genome datasets can improve our estimates of relatedness and pedigrees in captive breeding programs. The massive scale of whole genomes allows us to more easily trace the genealogical history of individuals, allowing us to assign parents more accurately. This also helps with our estimations of genetic relatedness, arguably the most critical aspect of genetic-based breeding programs. This is particularly helpful for species with tricky mating patterns, such as polyamory, brood spawning or difficult to track organisms.

The way forwards
While many non-model species are still lacking in the available genomic information, whole genomes are progressively being sequenced for more and more species. As this astronomical dataset grows, our ability to investigate, discover and test theories about evolution, natural selection and conservation will also improve. Many projects already exist which aim specifically to increase the number of whole genomes available for certain taxonomic groups such as birds and bats: these will no doubt prove to be invaluable resources for future studies.