It shouldn’t come as a surprise to anyone with a basic understanding of evolution that it is a temporal (and also spatial concept). Time is a fundamental aspect of the process of evolution by natural selection, and without it evolution wouldn’t exist. But time is also a fickle thing, and although it remains constant (let’s not delve into that issue here) not all things experience it in the same way.
This is based on the idea that for genes that are not related to traits under selection (either positively or negatively), new mutations should be acquired and lost under predominantly random patterns. Although this accumulation of mutations is influenced to some degree by alternate factors such as population size, the overall average of a genome should give a picture that largely discounts natural selection. But is this true? Is the genome truly neutral if averaged?
Non-neutrality
First, let’s take a look at what we mean by neutral or not. For genes that are not under selection, alleles should be maintained at approximately balanced frequencies and all non-adaptive genes across the genome should have relatively similar distribution of frequencies. While natural selection is one obvious way allele frequencies can be altered (either favourably or detrimentally), other factors can play a role.
An example of how linkage disequilibrium can alter allele frequency of ‘neutral’ parts of the genome as well. In this example, only one part of this section of the genome is selected for: the green gene. Because of this positive selection, the frequency of a particular allele at this gene increases (the blue graph): however, nearby parts of the genome also increase in frequency due to their proximity to this selected gene, which decreases with distance. The extent of this effect determines the size of the ‘linkage block’ (see below).
Why might ‘neutral’ models not be neutral?
The assumption that the vast majority of the genome evolves under neutral patterns has long underpinned many concepts of population and evolutionary genetics. But it’s never been all that clear exactly how much of the genome is actually evolving neutrally or adaptively. How far natural selection reaches beyond a single gene under selection depends on a few different factors: let’s take a look at a few of them.
Linked selection
As described above, physically close genes (i.e. located near one another on a chromosome) often share some impacts of selection due to reduced recombination that occurs at that part of the genome. In this case, even alleles that are not adaptive (or maladaptive) may have altered frequencies simply due to their proximity to a gene that is under selection (either positive or negative).
A (perhaps familiar) example of the interaction between recombination (the breaking and mixing of different genes across chromosomes) and linkage disequilibrium. In this example, we have 5 different copies of a part of the genome (different coloured sequences), which we randomly ‘break’ into separate fragments (breaks indicated by the dashed lines). If we focus on a particular base in the sequence (the yellow A) and count the number of times a particular base pair is on the same fragment, we can see how physically close bases are more likely to be coinherited than further ones (bottom column graph). This makes mathematical sense: if two bases are further apart, you’re more likely to have a break that separates them. This is the very basic underpinning of linkage and recombination, and the size of the region where bases are likely to be coinherited is called the ‘linkage block’.
The extent of this linkage effect depends on a number of other factors such as ploidy (the number of copies of a chromosome a species has), the size of the population and the strength of selection around the central locus. The presence of linkage and its impact on the distribution of genetic diversity (LD) has been well documented within evolutionary and ecological genetic literature. The more pressing question is one of extent: how much of the genome has been impacted by linkage? Is any of the genome unaffected by the process?
A cartoonish example of how background selection affects neighbouring sections of the genome. In this example, we have 4 genes (A, B, C and D) with interspersing neutral ‘non-gene’ sections. The allele for Gene B is strongly selected againstby natural selection (depicted here as the Banhammer of Selection). However, the Banhammer is not very precise, and when decreasing the frequency of this maladaptive Gene B allele it also knocks down the neighbouring non-gene sections. Despite themselves not being maladaptive, their allele frequencies are decreased due to physical linkage to Gene B.
This findings have significant implications for our understanding of the process of evolution, and how we can detect adaptation within the genome. In light of this research, there has been heated discussion about whether or not neutral theory is ‘dead’, or a useful concept.
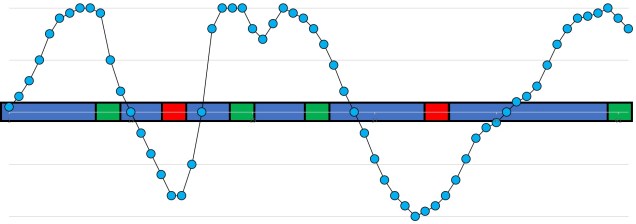
A vague summary of how a large portion of the genome might not actually be neutral. In this section of the genome, we have neutral (blue), maladaptive (red) and adaptive (green) elements. Natural selection either favours, disfavours, or is ambivalent about each of this sections alone. However, there is significant ‘spill-over’ around regions of positively or negatively selected sections, which causes the allele frequency of even the neutral sections to fluctuate widely. The blue dotted line represents this: when the line is above the genome, allele frequency is increased; when it is below it is decreased. As we travel along this section of the genome, you may notice it is rarely ever in the middle (the so-called ‘neutral‘ allele frequency, in line with the genome).
Although I avoid having a strong stance here (if you’re an evolutionary geneticist yourself, I will allow you to draw your own conclusions), it is my belief that the model of neutral theory – and the methods that rely upon it – are still fundamental to our understanding of evolution. Although it may present itself as a more conservative way to identify adaptation within the genome, and cannot account for the effect of the above processes, neutral theory undoubtedly presents itself as a direct and well-implemented strategy to understand adaptation and demography.
The classic way for new genetic variants to appear is often thought of as mutation: changes in a single base in the DNA are caused by various external processes such as chemical, physical or environmental influences (such as the sci-fi classics like UV rays or toxic chemicals). Although these forms of mutations happen very rarely and certainly don’t have the same effects comic books would leave you to believe, new mutations can occur relatively rapidly depending on the characteristics of the species. However, the most common way for new mutations to occur is actually part of the DNA replication process: copying DNA is not always perfect and even though the relevant proteins essentially run a spellcheck, sometimes the copy is not 100% perfect and new mutations occur.
An example of how adaptation can occur from a new mutation. In this example, we have one gene (TTXTT), with initial only one allele (variant), TTATT. In the second generation (row), a mutation occurs in one individual which creates a new, second allele: TTGTT. This allele is favoured over the TTATT allele, and in the next generation it’s frequency increases as the alternative allele frequency decreases (the pattern is shown in the frequency values on the right side).
Alternatively, genetic variation might already be present within a species or population. This is more likely if population sizes are large and populations are well connected and interbreeding. We refer to this diverse initial gene pool as ‘standing genetic variation’: that is, the amount of genetic variation within the population or species before the selective pressure requiring adaptation. Standing genetic variation can be thought of as the ‘diversity of choices’ for natural selection to act upon: the variants are readily available, and if a good choice exists it will be favoured by natural selection and become more widespread within the population or species (i.e. evolve).
A slightly more complex example of how adaptation can occur from standing variation, this time with two different genes. One codes for fur colour, with two different alleles: GCATA codes for orange fur, and GCGTA codes for grey fur. The other gene codes for ear tufts, with TTCCT coding for tufts and TCCCT coding for no tufts. Natural selection favours both orange fur and tufted ears, and cats with these traits reproduce more frequently than those without (see graph below). These cats probably look familiar.The frequency of all four alleles (i.e. either allele for both genes) over the generations in the above figure. Clearly, we can see how adaptation rapidly favours orange fur and tufted ears over grey fur and non-tufted ears with the shifts in frequencies over the different alleles.
We’ve discussed standing genetic variation before on The G-CAT, but often in a different light (and phrasing). For example, when we’ve talked about founder effect: that is, when a population is formed from only a few different individuals which causes it to be very genetically depauperate. In populations under strong founder effect, there is very little standing genetic variation for natural selection to act upon. This has long been an enigma for many pest species: how have they managed to proliferate so widely when they often originate from so few individuals and lack genetic diversity?
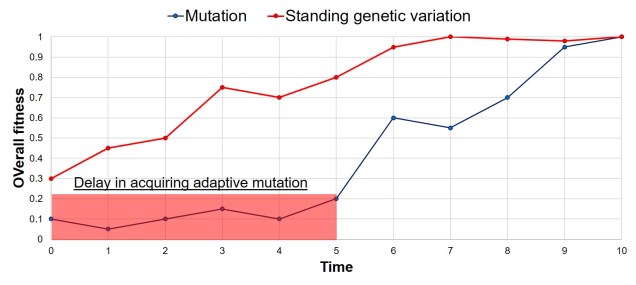
A rough example of the speed of adaptation depending on how the adaptive allele originated: whether it was already present (in the form of standing variation), or whether it was created by a new mutation. As one would expect, there is a significant lag delay in adaptation in the mutation scenario, based on the time it takes for said adaptive mutation to be created through relatively random processes. Thus, a positively selected allele from standing variation can allow a species to adapt much faster than waiting for a positive mutation to occur.
An extreme example of alternate splicing of one gene. We start with a single gene, composed of 5 (A–E) main gene elements (exons). Different environmental pressures (like fire risk, flooding, cold weather or predators, for example) cause the organism to use different combinations of these exons to make different proteins (right side; A–D). Actual alternate splicing is not usually this straight-forward (one gene doesn’t conveniently split into four forms depending on the threat), but the process is generally the same.