We’ve talked previously on The G-CAT about how the genetic underpinning of certain evolutionary traits can change in different directions depending on the selective pressure it is under. Particularly, we can see how the frequency of different alleles might change in one direction or another, or stabilise somewhere in the middle, depending on its encoded trait. But thinking bigger picture than just the genetics of one trait, we can actually see that evolution as an entire process works rather similarly.
Divergent evolution
The classic view of the direction of evolution is based on divergent evolution. This is simply the idea that a particular species possess some ancestraltrait. The species (or population) then splits into two (for one reason or another), and each one of these resultant species and populations evolves in a different way to the other. Over time, this means that their traits are changing in different directions, but ultimately originate from the same ancestral source.
Evidence for divergent evolution is rife throughout nature, and is a fundamental component of all of our understanding of evolution. Divergent evolution means that, by comparing similar traits in two species (called homologous traits), we can trace back species histories to common ancestors. Some impressive examples of this exist in nature, such as the number of bones in most mammalian species. Humans have the same number of neck bones as giraffes; thus, we can suggest that the ancestor of both species (and all mammals) probably had a similar number of neck bones. It’s just that the giraffe lineage evolved longer bones whereas other lineages did not.
A diagrammatic example of homologous structures in ‘hand’ bones. The coloured bones demonstrate how the same original bone structures have diverged into different forms. Source: BiologyWise.
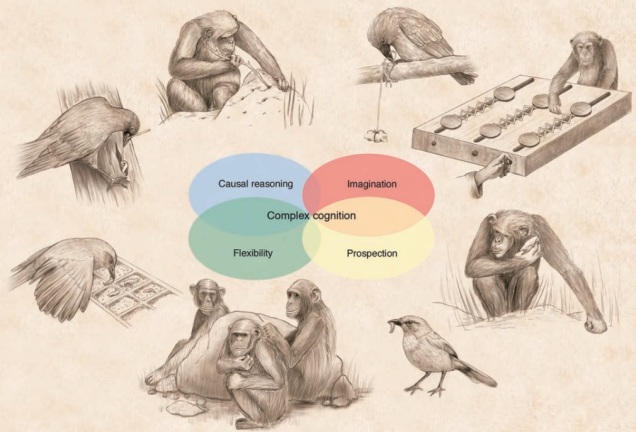
A surprising example of convergent evolution is cognitive ability in apes and some bird groups (e.g. corvids). There’s plenty of other animal groups more related to each of these that don’t demonstrate the same level of cognitive reasoning (based on the traits listed in the centre): thus, we can conclude that cognition has evolved twice in very, very different lineages. Source: Emery & Clayton, 2004.
A more dramatic (and potentially obvious) example of convergent evolution would be wings and the power of flight. Despite the fact that butterflies, bees, birds and bats all have wings and can fly, most of them are pretty unrelated to one another. It seems much more likely that flight evolved independently multiple times, rather than the other 99% of species that shared the same ancestor lost the capacity of flight.
Parallel evolution is an interesting field of research for a few reasons. Firstly, it provides a scenario in which we can more rigorously test expectations and outcomes of evolution in a particular environment. For example, if we find traits that are parallel in a whole bunch of fish species in a particular region, we can start to look at how that particular environment drives evolution across all fish species, as opposed to one species case studies.
Here’s another weird example; different populations of marsupials (particularly kangaroos and wallabies) show preferential handedness depending on where the population is. That is, different populations of different species of marsupials shows parallel evolution of handedness, since they’re related to one another but have evolved it independently of the other species. Source: Giljov et al. (2015).
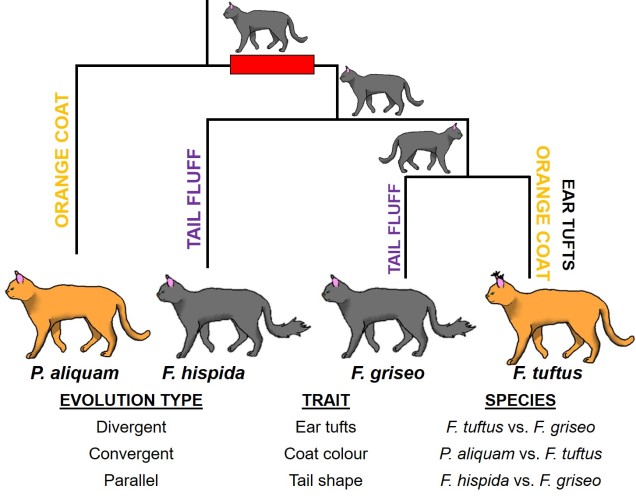
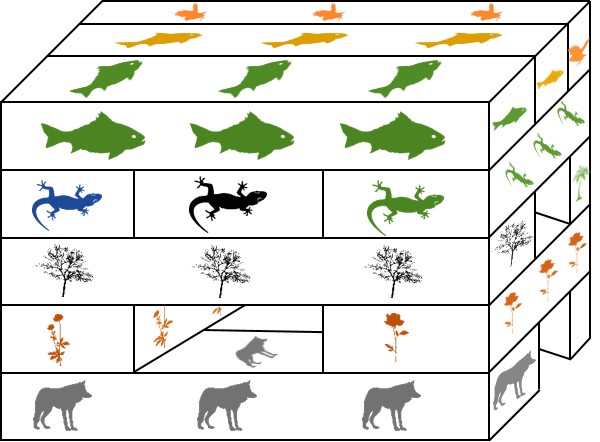
An example of all three types of evolutionary trajectory in a single phylogeny of cats (you know how we do it here at The G-CAT). This phylogeny consists of two distinct genera; one with one species (P. aliquam) and another of three species (the red box indicates their distance). Our species have three main physical traits: coat colour, ear tufts and tail shape. At the ancestral nodes of the tree, we can see what the ancestor of these species looked like for these three traits. Each of these traits has undergone a different type of evolution. The tufts on the ears are the result of divergent evolution, since F. tuftus evolved the trait differently to its nearest relative, F. griseo. Contrastingly, the orange coat colour of F. tuftus and P. aliquam are the result of convergent evolution: neither of these species are very closely related (remembering the red box) and evolved orange coats independently of one another (since their ancestors are grey). And finally, the fluffy tails of F. hispida and F. griseo can be considered parallel evolution, since they’re similar evolutionarily (same genus) but still each evolved tail fluff independently (not in the ancestor). This example is a little convoluted, but if you trace the history of each trait in the phylogeny you can more easily see these different patterns.
So, where is evolution going for nature? Well, the answer is probably all over the place, but steered by the current environmental circumstances. Predicting the evolutionary impacts of particular environmental change (e.g. climate change) is exceedingly difficult but a critical component of understanding the process of evolution and the future of species. Evolution continually surprises us with creative solution to complex problems and I have no doubt new mysteries will continue to be thrown at us as we delve deeper.
If bigger is better, then biggest is best. Having the genome of a particular study species fully sequenced allows us to potentially look at all of the genetic variation in the entire gene pool: but how do we sequence the entirety of the genome? And what are the benefits of having a whole genome to refer to?
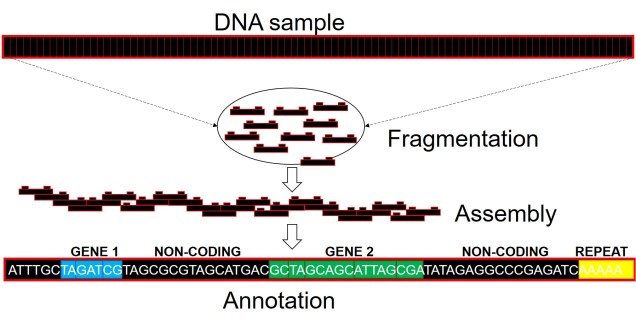
A very, very simplified overview of whole genome sequencing. Similar to other genomic technologies, we start by fragmenting the genome into much smaller, easier to sequence parts (reads). We then use a computer algorithm which pieces these reads together into a consecutive sequence based on overlapping DNA sequence (like building a chain out of Lego blocks). From this assembled genome, we can then attach annotations using information from other species’ genomes or genetic studies, which can correlate a particular sequence to a gene, a function of that gene, and the resultant protein from these gene (although not always are all of these aspects included).
Well, assembling the whole genome of an organism for the first time is a very tricky process. It involves taking DNA sequence from only a few individuals, breaking them down into smaller fragments and multiplying these fragments into the billions (moreorless the same process used in other genomics technologies: the real difference is that we need the full breadth of the genome so that we don’t miss any spaces). From these fragments, we use a complex computer algorithm which builds up a consensus sequence like a Lego tower; by finding parts of sequences which overlap, the software figures out which pieces connect to one another. Hopefully, we eventually end up with one very long continuous sequence; the genome! Sometimes, we might end with a few very large blocks (called contigs), but this is also useful for analyses (correlated with how many/big blocks there are). With this full genome, we use information from other more completed genomes (such as those from model species like humans, mice or even worms) to figure out which sections of the genome relate to specific genes. We can then annotate these sections by labelling them as clear genes, complete with start and end point, and attach a particular physical function of that gene.
The benefits of whole genomes
Having an entire genome as a reference is an extremely helpful tool in conservation and evolutionary studies. The first, and perhaps most obvious benefit, is the sheer scale of the data we can use. By having the entirety of the genome available, we can use potentially billions of base pairs of sequence in our genetic analyses (for reference, the human genome is >3 billion base pairs long). Even if we don’t sequence the full genome for all of our samples, having a reference genome as basis for assembly our reduced datasets significantly improves the quantity and quality of sequences we can use.
Another very important benefit is the ability to prescribe function in our studies. Many of our processes for obtaining data, even for genomic technologies, use random and anonymous fragments of the genome. Although this is a cost-effective way to obtain a very large amount of data, it unfortunately means that we often have no idea which part of the genome our sequences came from. This means that we don’t know which sequences relate to specific genes, and even if we did we would have no idea what those genes are or do! But with an annotated genome, we can take even our fragmented sequence and check it against the genome and find out what genes are present.
Understanding adaptation
Based on that, it seems pretty obvious about exactly how having an annotated genome can help us in studies of adaptation. Knowing the functional aspect of our genetic data allows us to more directly determine how evolution is happening in nature; instead of only being able to say that two species are evolving differently from one another, for example, we can explicitly look at how they are evolving. Is one evolving tolerance to hotter temperatures? Are they evolving different genes to handle different diets? Are they evolving in response to an external influence, like a viral outbreak or changing climate? What are the physiological consequences of these changes? These questions are critical in understanding past and future evolution, and full genome analysis allows us to delve into them much deeper.
A (slightly edited) figure of full genome comparisons between domestic dogs and wild wolves by Axelsson et al. (2013), with the aim of understanding the evolutionary changes associated with domestication. For avid readers, this figure probably looks familiar. This figure compares the genetic differentiation across the entire genome between dogs and wolves, with some sections of the genome (circled) showing clear differences. As there is an annotated dog genome, the authors then delved into these genes to understand the functional differences between the two. By comparing their genetic differences to functional genes, the authors can more explicitly suggest mechanisms or changes associated with the domestication process (such as adaptation to a starch-heavy and human-influenced diet).
This includes allowing us to better understand how adaptation actually works in nature. As we’ve discussed before, more traditional studies often assumed that single, or very few, genes were responsible for allowing a species to adapt and change, and that these genes had very strong effects on their physiology. But what we see far more often is polygenic adaptation; small changes in a very large number of genes which, combined together, allow the species to adapt and evolve. By having the entirety of the genome available, we are much more likely to capture all of the genes that are under natural selection in a particular population or species, painting a clearer picture of their evolutionary trajectory.
An example of linkage as a process. We start with a particular sequence (top); during recombination, this sequence may randomly break and rearrange into different parts. In this example, I’ve simulated four different ‘breaks’ (dashed coloured lines) due to recombination. Each of these breaks leads to two separate blocks of fragments; for example, the break at the blue line results in the second two sequence blocks (middle). If we focus on one target base pair in the sequence (golden A), then we can see in some fragments it remains with certain bases, but sometimes it gets separated by the break. If we compare how often the golden A is in the same block (i.e. is co-inherited) as each of the other bases, across all 4 breaks, then we see that the bases that are closest to it (the golden A is represented by the golden bar) are almost always in the same block. This makes sense: the further away a base is from our target, the more likely that there will be a break between it. This is shown in the frequency distributions at the bottom: the left figure shows the actual frequencies of co-inheritance (i.e. linkage) using the top example and those 4 breaks. The right figure shows a more realistic depiction of how linkage looks in the genome; it rapidly decays as we move away from the target (although the width and rate of this can vary).
An example of how whole genomes can improve our estimation of pedigrees. Say we have a random individual (star), and we want to know how they fit into a particular family tree (pedigree). With only a few genes, we might struggle to pick where in the family it fits based on limited genetic information. With a larger genetic dataset (such as reduced-representation genomics), we might be able to cross off a few potential candidate spots but still have some trouble with a few places (due to unknown parents, polygamy or issues with genetic analysis). With whole genomes, we should be able to much better clarify the whole pedigree and find exactly where our star individual fits in the tree (red circle). It is thanks to whole genomes, we can do those ancestry analyses that have gone viral lately!
The way forwards
While many non-model species are still lacking in the available genomic information, whole genomes are progressively being sequenced for more and more species. As this astronomical dataset grows, our ability to investigate, discover and test theories about evolution, natural selection and conservation will also improve. Many projects already exist which aim specifically to increase the number of whole genomes available for certain taxonomic groups such as birds and bats: these will no doubt prove to be invaluable resources for future studies.
‘Diversity’ is a term that gets used a lot these days, albeit usually in reference to social changes and structures. However, diversity is not merely a human construct and reflects an extremely important aspect of the natural world at a variety of levels. From the smallest genes to the biggest ecosystems, diversity is a trait that confers a massive range of benefits to individuals, populations, species and even the entire globe. Let’s dissect this diversity down at different scales and see how beneficial it can be.
The generalised hierarchy at life, with diversity being an important component of each tier. At the smallest tier, genes underpin all life. The collection of genetic diversity is often summarised into a population (as a single cohesive genetic unit). Several populations can be pooled together into a single (usually) cohesive species. Different species are then components of a larger community (which in turn are components of a broader ecosystem).
Genetic diversity
At the smallest scale in the hierarchy of genetic differentiation, we have the genes themselves. It is a well-established concept that having a diversity of genetic variants (alleles) within a population or species is critical to their future adaptation, evolution and persistance. This is because different alleles will have different benefits (or costs) depending on the environmental pressure that influences them; natural selection might favour one allele over another at one time, but a different one as the pressure changes. Having a higher number of alleles within the population or species means that there is a greater chance at least a few individuals will possess an adaptive gene with the changing environment (which we know can be quite rapid and very, very strong). The diversity serves as a ‘buffer’ against extinction; evolution by natural selection functions best when there are many options to choose from.
Without this diversity, species run the risk of having no adaptive genes at the ready to deal with a selective pressure. Either a new adaptive gene must mutate (or come about in other ways, such as through gene flow from another population or species) or the population/species will suffer and potentially go extinct. As strong selection causes the species to dwindle, it enters what is referred to as the ‘extinction vortex’. Without genetic diversity, they can’t adapt: thus, more individuals die off, causing more genetic diversity to be lost from the population. This pattern is a vicious cycle which can inevitably destroy species (without serious intervention).
A very dramatic representation of the extinction vortex.
For this reason, captive breeding programs aim to maintain as much of the genetic diversity of the original population as possible. This reduces the probability of entering a downward extinction spiral from inbreeding depression and helps to maintain populations into the future (both the captive one and the wild population when we reintroduce individuals into the wild).
“Population” diversity
Because genetic diversity is critically important for species survival, we must also try to preserve the diversity of the entire gene pool of a species. This means conserving highly genetically differentiated populations within a species as a priority, as they may be the only ones that possess the necessary adaptive genes to save the rest of the species. This adaptive genetic variation can then be introduced into other populations in genetic rescue programs and serve as a means to semi-naturally allow the species to evolve. Evolutionarily-significant units (ESUs) are one measure of the invaluable nature of genetically unique populations.
Although many more traditional conservationists strongly believe that ESUs should be managed entirely independently of one another (to preserve their evolutionary ‘pedigree’ and prevent the risk of outbreeding depression), it has been suggested that the benefit of genetic rescue in many cases significantly outweighs this risk of outbreeding depression. For some species, this really is an act of rescue: they are at the edge of extinction, and if we do nothing we condemn them to die out.
Introducing genetic material across populations (or even species!) can generate new functional genes that allow the recipient species to adapt to selective pressures. This might sound very strange, and could be extremely rare, but examples of adaptive genetic material in one species originating from another species through hybridisation do exist in nature. For example, the black coat of wolves is a highly adaptive trait in some populations and is encoded for by the Melanocortin 1 receptor (Mc1r) gene. However, the specific mutation in Mc1r gene that generates the black coat colour actually first originated in domestic dogs; when wild wolves and domestic dogs interbred, this mutation was transferred into the wolf gene pool. Natural selection strongly favoured this new variant, and it very rapidly underwent strong positive selection. Thus, the adaptiveness of black wolves is thanks to a domestic dog mutation!
Species diversity
At a higher level of the hierarchy, the diversity of species within a particular community or ecosystem has been shown to be important for the health and stability of said community. Every species, however small or seemingly unimpressive, plays a role in the greater ecosystem balance, through interactions with other species (e.g. as predator, as prey, as competitor) and the abiotic environment. While some species are known to have very strong impacts on the immediate ecosystem (often dubbed ‘keystone species’, such as apex predators), all species have some influence on the world around them (we’re especially good at it).
The overall health and stability of an ecosystem, as well as the benefits it can provide to all living things (including humans) is largely determined by the diversity of species. For example, ‘habitat engineers’ are types of species that, by altering the physical environment around them (such as to build a home), directly provide new habitat for other species. They are a fundamental underpinning of many incredibly vibrant ecosystems; think of what a reef system would look like if there were no corals in it. There’d be no anemones growing colourfully; no fish to live in them; no sharks to feed on these non-existent fish. This is just one example of a complex ecosystem that truly relies on its inhabiting species to function.
Much like Jenga, taking out one block (a species) could cause the entire stack (the ecosystem) to collapse in on itself. Even if it stands up, however, the system will still be weaker without the full diversity to support it.
Protecting our diversity
Diversity is not just a social construct and is an important phenomenon in nature, at a variety of different levels. Preserving the full diversity of life, from genetic diversity within populations and species to full species diversity within ecosystems, is critical to maintaining healthy and robust natural systems. The more diversity we have at each level of this hierarchy, the greater robustness and security we will have in the future.
The distribution of organisms across the Earth, both over time and across space, is a fundamental aspect of the field of biogeography. But our understanding of the mechanisms by which organisms are distributed across the globe, and how this affects their evolution, can be at times highly enigmatic. Why are Australia and the Americas the only two places that have marsupials? How did lemurs get all the way to Madagascar, and why are they the only primate that has made the trip? How did Darwin’s famous finches get over to the Galápagos, and why are there so many species of them there now?
All of these questions can be addressed with a combination of genetic, environmental and ecological information across a variety of timescales. However, the overall field of biogeography (and phylogeography as a derivative of it) has traditionally been largely rooted on a strong yet changing theoretical basis. The earliest discussions and discoveries related to biogeography as a field of science date back to the 18th Century, and to Carl Linnaeus (to whom we owe our binomial classification system) and Alexander von Humboldt. These scientists (and undoubtedly many others of that era) were among the first to notice how organisms in similar climates (e.g. Australia, South Africa and South America) showed similar physical characteristics despite being so distantly separated (both in their groups and geographic distance). The communities of these regions also appeared to be highly similar. So how could this be possible over such huge distances?
A pretty unreasonable mechanism (and example) of dispersal in foxes. And yes, all tourists wear sunglasses and Hawaiian shirts, even arctic fox ones.
Dispersal or vicariance?
Two main explanations for these patterns are possible; dispersal and vicariance. As one might expect, dispersal denotes that an ancestral species was distributed in one of these places (referred to as the ‘centre of origin’) before it migrated and inhabited the other places. Contrastingly, vicariancesuggests that the ancestral species was distributed everywhere originally, covering all contemporary ranges within it. However, changes in geography, climate or the formation of other barriers caused the range of the ancestor to fragment, with each fragmented group evolving into its own distinct species (or group of species).
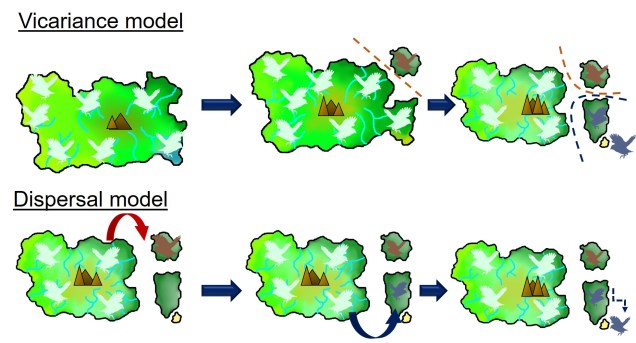
An example of dispersal vs. vicariance patterns of biogeography in an island bird (pale blue). In the top example, the sequential separation of parts of the island also cause parts of the distribution of the original bird species to become fragmented. These fragments each evolve independently of their ancestor and form new species (red, and then blue). In the bottom example, the island geography doesn’t change but in rare events a bird disperses from the main island onto a new island. The new selective pressures of that island cause the dispersed birds to evolve into new species (red and blue). In both examples, islands that were recently connected or are easy to disperse across do not generate new species (in the sandy island in the bottom right). You’ll notice that both processes result in the same biogeographic distribution of species.
In initial biogeographic science, dispersal was the most heavily favoured explanation. At the time, there was no clear mechanism by which organisms could be present all over the globe without some form of dispersal: it was generally believed that the world was a static, unmoving system. Dispersal was well supported by some biological evidence such as the diversification of Darwin’s finches across the Galápagos archipelago. Thus, this concept was supported through the proposals of a number of prominent scientists such as Charles Darwin and A.R. Wallace. For others, however, the distance required for dispersal (such as across entire oceans) seemed implausible and biologically unrealistic.
A paradigm shift in biogeography
Two particular developments in theory are credited with a paradigm shift in the field; cladistics and plate tectonics. Cladistics simply involved using shared biological characteristics to reconstruct the evolutionary relationships of species (think like phylogenetics, but using physical traits instead of genetic sequence). Just as importantly, however, was plate tectonic theory, which provided a clear way for organisms to spread across the planet. By understanding that, deep in the past, all continents had been directly connected to one another provides a convenient explanation for how species groups spread. Instead of requiring for species to travel across entire oceans, continental drift meant that one widespread and ancient ancestor on the historic supercontinent (Pangaea; or subsequently Gondwana and Laurasia) could become fragmented. It only required that groups were very old, but not necessarily very dispersive.
Surf’s up, dudes! Although continental drift was no doubt an important factor in the distribution and dispersal of many organisms on Earth, it actually probably wasn’t the reason lemurs got to Madagascar. Sorry for the mislead.
From these advances in theory, cladistic vicariance biogeography was born. The field rapidly overtook dispersal as the most likely explanation for biogeographic patterns across the globe by not only providing a clear mechanism to explain these but also an analytical framework to test questions relating to these patterns. Further developments into the analytical backbone of cladistic vicariance allowed for more nuanced questions of biogeography to be asked, although still fundamentally ignored the role of potential dispersals in explaining species’ distributions.
Modern philosophy of biogeography
So, what is the current state of the field? Well, the more we research biogeographic patterns with better data (such as with genomics) the more we realise just how complicated the history of life on Earth can be. Complex modelling (such as Bayesian methods) allow us to more explicitly test the impact of Earth history events on our study species, and can provide more detailed overview of the evolutionary history of the species (such as by directly estimating times of divergence, amount of dispersal, extent of range shifts).
From a theoretical perspective, the consistency of patterns of groups is always in question and exactly what determines what species occurs where is still somewhat debatable. However, the greater number of types of data we can now include (such as geological, paleontological, climatic, hydrological, genetic…the list goes on!) allows us to paint a better picture of life on Earth. By combining information about what we know happened on Earth, with what we know has happened to species, we can start to make links between Earth history and species history to better understand how (or if) these events have shaped evolution.
Understanding the evolutionary history of species can be a complicated matter, both from theoretical and analytical perspectives. Although phylogenetics addresses many questions about evolutionary history, there are a number of limitations we need to consider in our interpretations.
One of these limitations we often want to explore in better detail is the estimation of the divergence times within the phylogeny; we want to know exactly when two evolutionary lineages (be they genera, species or populations) separated from one another. This is particularly important if we want to relate these divergences to Earth history and environmental factors to better understand the driving forces behind evolution and speciation. A traditional phylogenetic tree, however, won’t show this: the tree is scaled in terms of the genetic differences between the different samples in the tree. The rate of genetic differentiation is not always a linear relationship with time and definitely doesn’t appear to be universal.
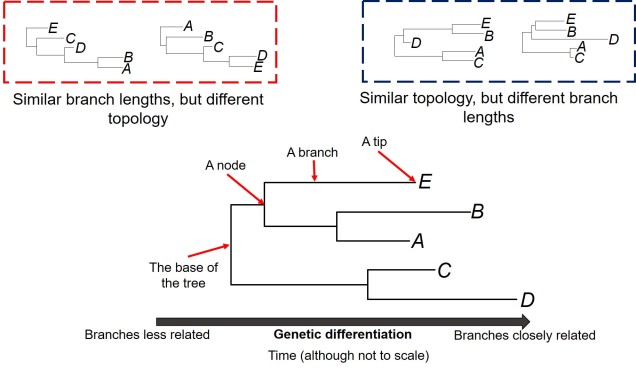
The general anatomy of a phylogenetic tree. A phylogeny describes the relationships of tips (i.e. which are more closely related than others; referred to as the topology), how different these tips are (the length of the branches) and the order they separated in time (separations shown by the nodes). Different trees can share some traits but not others: the red box shows two phylogenetic trees with similar branch lengths (all of the branches are roughly the same) but different topology (the tips connect differently: A and B are together on the left but not on the right, for example). Conversely, two trees can have the same topology, but show differing lengths in the branches of the same tree (blue box). Note that the tips are all in the same positions in these two trees. Typically, it’s easier to read a tree from right to left: the two tips who have branches that meet first are most similar genetically; the longer it takes for two tips to meet along the branches, the less similar they are genetically.
How do we do it?
The parameters
There are a number of parameters that are required for estimating divergence times from a phylogenetic tree. These can be summarised into two distinct categories: the tree model and the substitution model.
The first one of these is relatively easy to explain; it describes the exact relationship of the different samples in our dataset (i.e. the phylogenetic tree). Naturally, this includes the topology of the tree (which determines which divergences times can be estimated for in the first place). However, there is another very important factor in the process: the lengths of the branches within the phylogenetic tree. Branch lengths are related to the amount of genetic differentiation between the different tips of the tree. The longer the branch, the more genetic differentiation that must have accumulated (and usually also meaning that longer time has occurred from one end of the branch to the other). Even two phylogenetic trees with identical topology can give very different results if they vary in their branch lengths (see the above Figure).
The second category determines how likely mutations are between one particular type of nucleotide and another. While the details of this can get very convoluted, it essentially determines how quickly we expect certain mutations to accumulate over time, which will inevitably alter our predictions of how much time has passed along any given branch of the tree.
Calibrating the tree
However, at least one another important component is necessary to turn divergence time estimates into absolute, objective times. An external factor with an attached date is needed to calibrate the relative branch divergences; this can be in the form of the determined mutation rate for all of the branches of the tree or by dating at least one node in the tree using additional information. These help to anchor either the mutation rate along the branches or the absolute date of at least one node in the tree (with the rest estimated relative to this point). The second method often involves placing a time constraint on a particular node of the tree based on prior information about the biogeography of the species (for example, we might know one species likely diverged from another after a mountain range formed: the age of the mountain range would be our constraints). Alternatively, we might include a fossil in the phylogeny which has been radiocarbon dated and place an absolute age on that instead.
Don’t you know it’s rude to ask an ammomite her age?
In regards to the former method, mutation rates describe how fast genetic differentiation accumulates as evolution occurs along the branch. Although mutations gradually accumulate over time, the rate at which they occur can depend on a variety of factors (even including the environment of the organism). Even within the genome of a single organism, there can be variation in the mutation rate: genes, for example, often gain mutations slower than non-coding region.
Although mutation rates (generally in the form of a ‘molecular clock’) have been traditionally used in smaller datasets (e.g. for mitochondrial DNA), there are inherent issues with its assumptions. One is that this rate will apply to all branches in a tree equally, when different branches may have different rates between them. Second, different parts of the genome (even within the same individual) will have different evolutionary rates (like genes vs. non-coding regions). Thus, we tend to prefer using calibrations from fossil data or based on biogeographic patterns (such as the time a barrier likely split two branches based on geological or climatic data).
The analytical framework
All of these components are combined into various analytical frameworks or programs, each of which handle the data in different ways. Many of these are Bayesian model-based analysis, which in short generates hypothetical models of evolutionary history and divergence times for the phylogeny and tests how well it fits the data provided (i.e. the phylogenetic tree). The algorithm then alters some aspect(s) of the model and tests whether this fits the data better than the previous model and repeats this for potentially millions of simulations to get the best model. Although models are typically a simplification of reality, they are a much more tractable approach to estimating divergence times (as well as a number of other types of evolutionary genetics analyses which incorporating modelling).
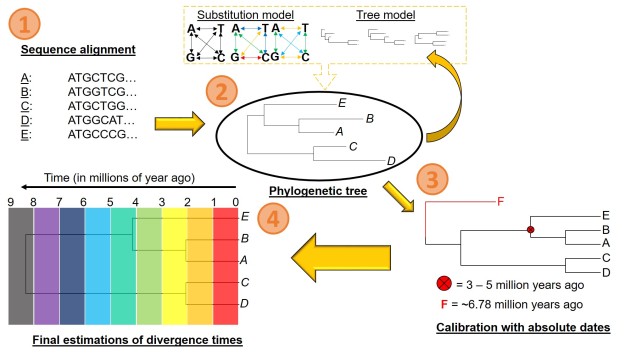
A (believe it or not, simplified) pipeline for estimating divergence times from a phylogeny. 1) We obtain our DNA sequences for our samples: in this example, we’ll see each Sample (A-E) is a representative of a single species. We align these together to make sure we’re comparing the same part of the genome across all of them. 2) We estimate the phylogenetic tree for our samples/species. In a Bayesian framework, this means creating simulation models containing a certain substitution model and a given tree model (containing certain topology and branch lengths). Together, these two models form the likelihood model: we then test how well this model explains our data (i.e. the likelihood of getting the patterns in our data if this model was true). We repeat these simulations potentially hundreds of thousands of times until we pinpoint the most likely model we can get. 3) Using our resulting phylogeny, we then calibrate some parts of it based on external information. This could either be by including a carbon-dated fossil (F) within the phylogeny, or constraining the age of one node based on biogeographic information (the red circle and cross). 4) Using these calibrations as a reference, we then estimated the most likely ages of all the splits in the tree, getting our final dated phylogeny.
Despite the developments in the analytical basis of estimating divergence times in the last few decades, there are still a number of limitations inherent in the process. Many of these relate to the assumptions of the underlying model (such as the correct and accurate phylogenetic tree and the correct estimations of evolutionary rate) used to build the analysis and generate simulations. In the case of calibrations, it is also critical that they are correctly dated based on independent methods: inaccurate radiocarbon dating of a fossil, for example, could throw out all of the estimations in the entire tree. That said, these factors are intrinsic to any phylogenetic analysis and regularly considered by evolutionary biologists in the interpretations and discussions of results (such as by including confidence intervals of estimations to demonstrate accuracy).
Understanding the temporal aspects of evolution and being able to relate them to a real estimate of age is a difficult affair, but an important component of many evolutionary studies. Obtaining good estimates of the timing of divergence of populations and species through molecular dating is but one aspect in building the picture of the history of all organisms, including (and especially) humans.