In the 18 years since the completion of the Human Genome Project, the practicality of assembling full genomes for a wide range of taxa beyond ourselves has only improved. While model taxa systems have achieved genomes before many others, it is now possible for whole genomes to be assembled for a range of non-model organisms as well. But how do we assemble the genome of a species for the very first time (often de novo – literally “from the new”)? What can we do with this genome? Why is it so useful? Let’s delve into the process and outcomes of genome assembly a little more.
If bigger is better, then biggest is best. Having the genome of a particular study species fully sequenced allows us to potentially look at all of the genetic variation in the entire gene pool: but how do we sequence the entirety of the genome? And what are the benefits of having a whole genome to refer to?
A very, very simplified overview of whole genome sequencing. Similar to other genomic technologies, we start by fragmenting the genome into much smaller, easier to sequence parts (reads). We then use a computer algorithm which pieces these reads together into a consecutive sequence based on overlapping DNA sequence (like building a chain out of Lego blocks). From this assembled genome, we can then attach annotations using information from other species’ genomes or genetic studies, which can correlate a particular sequence to a gene, a function of that gene, and the resultant protein from these gene (although not always are all of these aspects included).
Well, assembling the whole genome of an organism for the first time is a very tricky process. It involves taking DNA sequence from only a few individuals, breaking them down into smaller fragments and multiplying these fragments into the billions (moreorless the same process used in other genomics technologies: the real difference is that we need the full breadth of the genome so that we don’t miss any spaces). From these fragments, we use a complex computer algorithm which builds up a consensus sequence like a Lego tower; by finding parts of sequences which overlap, the software figures out which pieces connect to one another. Hopefully, we eventually end up with one very long continuous sequence; the genome! Sometimes, we might end with a few very large blocks (called contigs), but this is also useful for analyses (correlated with how many/big blocks there are). With this full genome, we use information from other more completed genomes (such as those from model species like humans, mice or even worms) to figure out which sections of the genome relate to specific genes. We can then annotate these sections by labelling them as clear genes, complete with start and end point, and attach a particular physical function of that gene.
The benefits of whole genomes
Having an entire genome as a reference is an extremely helpful tool in conservation and evolutionary studies. The first, and perhaps most obvious benefit, is the sheer scale of the data we can use. By having the entirety of the genome available, we can use potentially billions of base pairs of sequence in our genetic analyses (for reference, the human genome is >3 billion base pairs long). Even if we don’t sequence the full genome for all of our samples, having a reference genome as basis for assembly our reduced datasets significantly improves the quantity and quality of sequences we can use.
Another very important benefit is the ability to prescribe function in our studies. Many of our processes for obtaining data, even for genomic technologies, use random and anonymous fragments of the genome. Although this is a cost-effective way to obtain a very large amount of data, it unfortunately means that we often have no idea which part of the genome our sequences came from. This means that we don’t know which sequences relate to specific genes, and even if we did we would have no idea what those genes are or do! But with an annotated genome, we can take even our fragmented sequence and check it against the genome and find out what genes are present.
Understanding adaptation
Based on that, it seems pretty obvious about exactly how having an annotated genome can help us in studies of adaptation. Knowing the functional aspect of our genetic data allows us to more directly determine how evolution is happening in nature; instead of only being able to say that two species are evolving differently from one another, for example, we can explicitly look at how they are evolving. Is one evolving tolerance to hotter temperatures? Are they evolving different genes to handle different diets? Are they evolving in response to an external influence, like a viral outbreak or changing climate? What are the physiological consequences of these changes? These questions are critical in understanding past and future evolution, and full genome analysis allows us to delve into them much deeper.
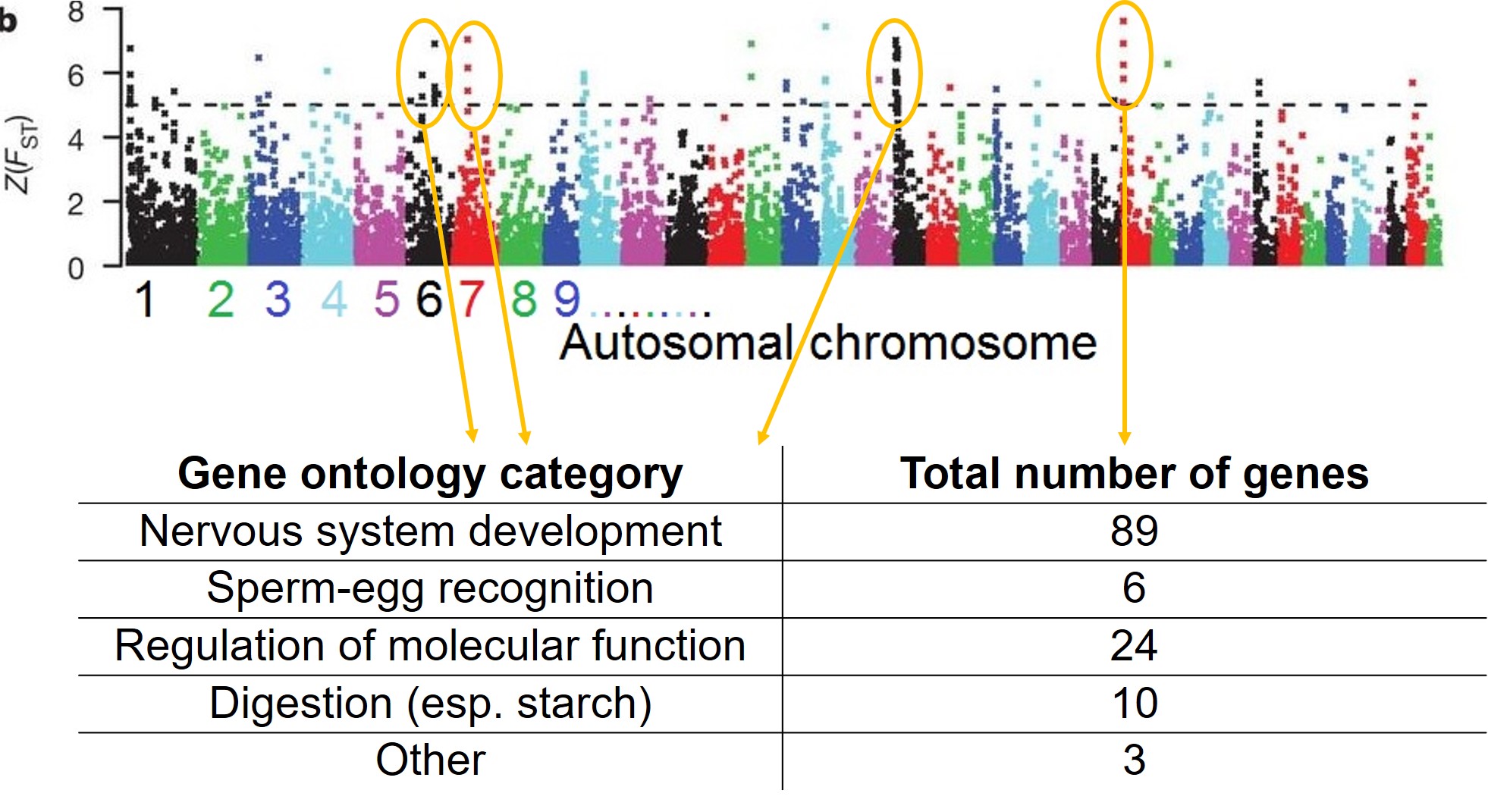
A (slightly edited) figure of full genome comparisons between domestic dogs and wild wolves by Axelsson et al. (2013), with the aim of understanding the evolutionary changes associated with domestication. For avid readers, this figure probably looks familiar. This figure compares the genetic differentiation across the entire genome between dogs and wolves, with some sections of the genome (circled) showing clear differences. As there is an annotated dog genome, the authors then delved into these genes to understand the functional differences between the two. By comparing their genetic differences to functional genes, the authors can more explicitly suggest mechanisms or changes associated with the domestication process (such as adaptation to a starch-heavy and human-influenced diet).
This includes allowing us to better understand how adaptation actually works in nature. As we’ve discussed before, more traditional studies often assumed that single, or very few, genes were responsible for allowing a species to adapt and change, and that these genes had very strong effects on their physiology. But what we see far more often is polygenic adaptation; small changes in a very large number of genes which, combined together, allow the species to adapt and evolve. By having the entirety of the genome available, we are much more likely to capture all of the genes that are under natural selection in a particular population or species, painting a clearer picture of their evolutionary trajectory.
An example of linkage as a process. We start with a particular sequence (top); during recombination, this sequence may randomly break and rearrange into different parts. In this example, I’ve simulated four different ‘breaks’ (dashed coloured lines) due to recombination. Each of these breaks leads to two separate blocks of fragments; for example, the break at the blue line results in the second two sequence blocks (middle). If we focus on one target base pair in the sequence (golden A), then we can see in some fragments it remains with certain bases, but sometimes it gets separated by the break. If we compare how often the golden A is in the same block (i.e. is co-inherited) as each of the other bases, across all 4 breaks, then we see that the bases that are closest to it (the golden A is represented by the golden bar) are almost always in the same block. This makes sense: the further away a base is from our target, the more likely that there will be a break between it. This is shown in the frequency distributions at the bottom: the left figure shows the actual frequencies of co-inheritance (i.e. linkage) using the top example and those 4 breaks. The right figure shows a more realistic depiction of how linkage looks in the genome; it rapidly decays as we move away from the target (although the width and rate of this can vary).
An example of how whole genomes can improve our estimation of pedigrees. Say we have a random individual (star), and we want to know how they fit into a particular family tree (pedigree). With only a few genes, we might struggle to pick where in the family it fits based on limited genetic information. With a larger genetic dataset (such as reduced-representation genomics), we might be able to cross off a few potential candidate spots but still have some trouble with a few places (due to unknown parents, polygamy or issues with genetic analysis). With whole genomes, we should be able to much better clarify the whole pedigree and find exactly where our star individual fits in the tree (red circle). It is thanks to whole genomes, we can do those ancestry analyses that have gone viral lately!
The way forwards
While many non-model species are still lacking in the available genomic information, whole genomes are progressively being sequenced for more and more species. As this astronomical dataset grows, our ability to investigate, discover and test theories about evolution, natural selection and conservation will also improve. Many projects already exist which aim specifically to increase the number of whole genomes available for certain taxonomic groups such as birds and bats: these will no doubt prove to be invaluable resources for future studies.