On the precipice
Species which exist in fragmented, isolated and reduced populations have elevated extinction risk. Not only are they more susceptible to demographic and environmental stochasticity, which can easily wipe out small populations, but they also suffer from a range of genetic impacts. Notably, populations often lose significant amounts of genetic diversity as they reduce in size, potentially losing important adaptive diversity enabling them to respond to current and future environmental change. At the same time, random genetic drift becomes stronger relative to natural selection, reducing the efficacy of selection to be able to increase the frequency of favourable alleles and reduce the frequency of maladaptive ones. Together, these impacts create feedback loops which hasten the decline into the extinction vortex.
Inbreeding and genetic bottlenecks
Another relevant factor is the increased degree of inbreeding in bottlenecked populations. With fewer individuals present in the population, mate choice becomes more limited and some individuals may need to breed with related individuals out of necessity. At the genetic level, “relatedness” refers to how genetically similar two individuals are (irrespective of pedigree-based relationship): the reduced genetic diversity of bottlenecked populations means that all potential pairings will be more “related” than in a larger population. Even if individuals can avoid mating with genealogically related mates (like siblings or parents), inbreeding may be unavoidable if even genealogically unrelated pairings are genetically related. Together, these drive an increase in homozygosity, one of the primary measures of population inbreeding. This elevated inbreeding can have significant impacts on the overall fitness and health of the population, and at the individual level, if adaptive variation is lost and maladaptive variation is boosted.
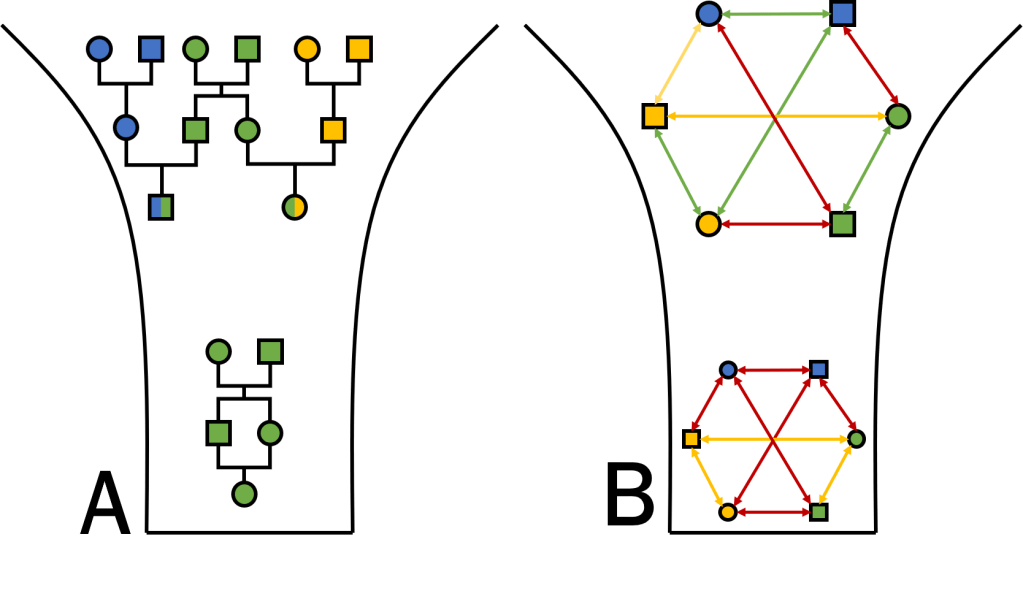
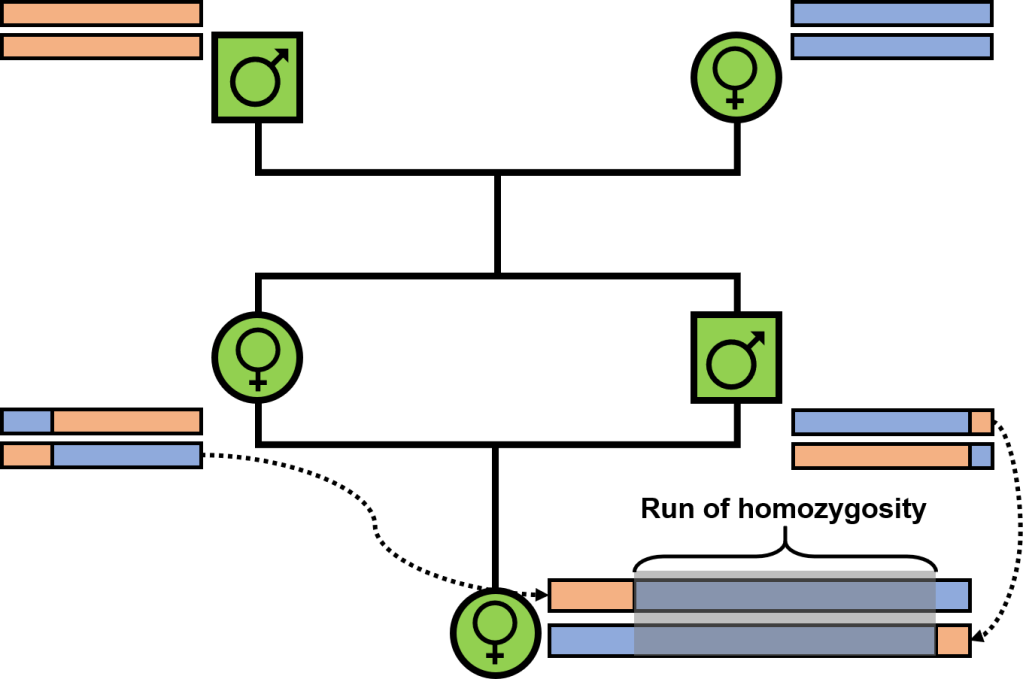
One of the more noticeable impacts of inbreeding is that inbred offspring will inherit larger sections of the genome which we call “identical-by-descent”: that is, both copies share identical variation because they originate in the recent past. In extreme inbreeding (between siblings), these segments may trace back to only a few generations (to their shared parents): in general, however, genomic segments will have variable degrees of ancestry depending on when new mutations arose and how recombination breaks up these segments to create new genetic combinations. Regardless, inbreeding inevitably creates large sections of the genome which are long contiguous sections of homozygous positions: even if there is variation in some of those positions at the population-level, there isn’t any differences between the two parents (because they are highly related) and so there is no variation within their offspring. Naturally, we call these “runs of homozygosity” (ROH), and they have some clear impacts on the fitness of individuals and populations.
The impacts of ROH
Aside from representing the loss of genetic diversity in bottlenecked populations, ROH have other, more tangible, negative impacts. Chiefly, ROH are likely to harbour an excess of deleterious mutations in homozygous form – we’ve (briefly) touched on these variants in the concept of “genetic load.” Many of these mutations may persist in populations over time due to being recessive and (often) only weakly deleterious: although present, their impact is hidden in heterozygous form (“masked”). However, when inbreeding drives a higher degree of homozygosity due to inheritance of identical-by-descent sequences, these deleterious mutations may be found in homozygous form (“realised”). Given that ROH comprise long segments of the genome which are identical-by-descent, it’s easy to see how these sections may contain an abundance of now-unmasked deleterious mutations, leading to some nasty impacts on fitness. In fact, the elevated exposure of deleterious mutations within ROH likely underpins many of the more dramatic impacts of inbreeding, including in humans and livestock. Avoiding large and long ROH is key to minimising the impact and outcome of inbreeding depression in small populations, which are already at higher risk of extinction.

But how can the damage of ROH be undone? Over time, long ROH may be broken up by recombination – when sections of the two pairs of chromosomes of an individual are mixed, creating new combinations of genetic variation. For ROH, this might mean separating two identical segments, reducing the size of the ROH eventually their total length relative to the genome of the organism. Exactly how fast this happens depends on several biological factors – not least of which is genetic diversity, which is already reduced in these inbred populations. But given enough time, recombination may effectively break down very long ROH into smaller and smaller fragments, reducing their impact on genetic diversity and the fitness of an individual. Introducing new genetic variation from another population – either by natural gene flow or through human-mediated genetic rescue – may significantly speed up the process as this new genetic material provides greater opportunity for heterozygous sites to break up the ROH.

Demography and ROH
Another neat outcome of this phenomenon is that we can actually use the relationship of recombination and ROH length to track the demographic history of a given population. Those that are only very recently inbred are expected to have significantly longer ROH which cover a greater expanse of the genome, since recombination hasn’t had time to dismantle the ROH yet. The further back in time this inbreeding occurred, however, the greater chance that recombination has shortened the ROH and thus the shorter average ROH length we might expect within the genome. The expected length of an ROH can thus be calculated (given a recombination rate, the number of these “crossing-over” events per generation; and time). Using these relationships and the known length of ROH across a given genome, we can trace their histories back in time to determine whether the inbreeding driving these patterns is recent (e.g., a result of human impacts) or more ancient (e.g., the result of past environmental changes), unlocking an important viewpoint of species histories.

In terms of conservation management, this may help us identify how severe the impact of inbreeding is – a lack of historical, but an abundance of recent, inbreeding might indicate that a species does not have the necessary time or biological mechanisms to deal with inbreeding depression caused by long ROH, prompting more immediate management actions such as genetic rescue or supervised captive breeding. In others, historical impacts of inbreeding might provide further context for management (in choosing which individuals to source for translocations, for example) or suggest that inbreeding depression is not an immediate threat (plenty of other things may be higher priority, though).
Running forwards
With the accelerating development of genomic sequencing technologies, more and more whole genome sequences for non-model species are becoming available. Detecting and measuring ROH across many individuals in endangered species allows us to better understand the nature of inbreeding depression and the risk this poses for their persistence and make management decisions accordingly. Furthermore, determining ROH across a greater variety of species allows us to delve more thoroughly into the mechanisms driving their origin or subsequent recombination: how do biological traits affect their proliferation? Do they always contribute strongly to negative fitness? What extent of ROH across the genome leads to significant inbreeding depression? These questions (and more) will only be revealed with more research into ROH dynamics and function, especially endangered or domesticated species.

One thought on “Runaway inbreeding: how runs of homozygosity impact conservation management”