From genotype to phenotype
One fundamental aspect of conservation and evolution research is the implicit connection between genetic variation, phenotypic characteristics, and their influence on Darwinian fitness. Genetic diversity underpins many aspects of the adaptive potential of a population, and many of the fundamental concepts of the field rely on the assumed connection between genetic and phenotypic characteristics. But this connection is neither straightforward, nor always predictable.
Functional genomics
Despite this well accepted association, understanding how genetic factors directly drive changes in physiology, behaviour, or other phenotypic traits – the true targets of natural selection – remains a challenge in most scenarios. But with the growing body of high-quality and annotated genomes available, it is now possible to make direct connections between genes, their resultant proteins, and the function they ultimately play in the physiology, behaviour, or other phenotypic traits of an organism. This aspect of biological science is referred to as “functional genomics” and plays an important role in many fields such as evolution, conservation, medicine, and psychology.
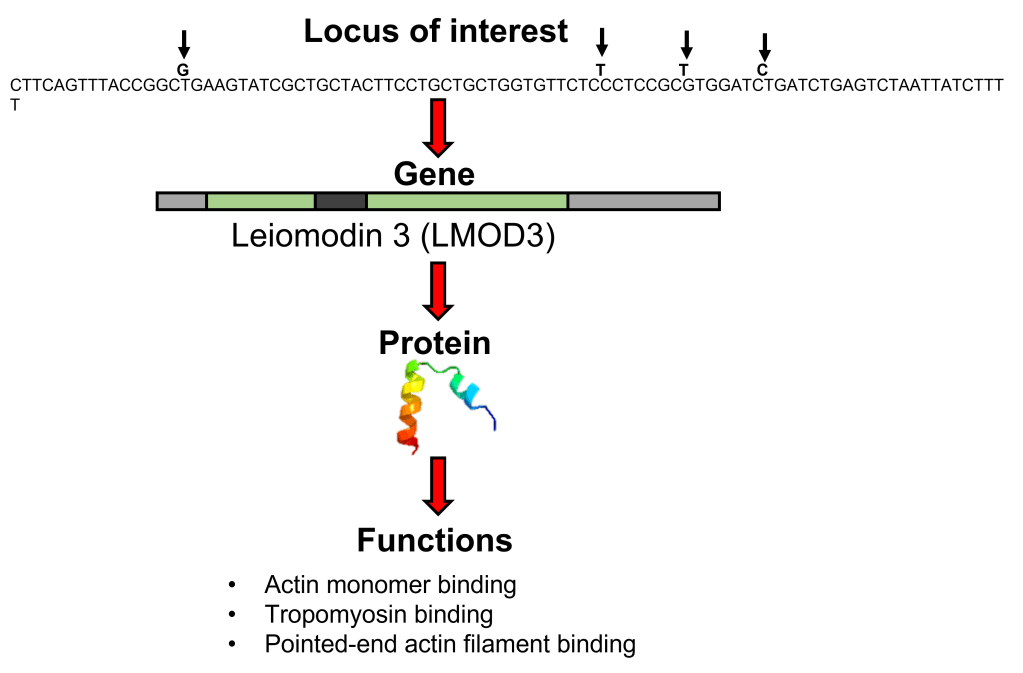
A (very) basic overview of the process of functional annotation, discussed in more detail here. This example is based on a locus of interest for one of my PhD chapters (as yet unpublished, in this part of the work will likely be removed for conciseness). A single locus (this one containing 4 SNPs (arrows)) is annotated using a genome, identifying the gene it occurs in. From here, we can determine the protein (note: the structure shown here is not actually what Leiomodin 3 looks like) which may have some identified biological functions.
Within an evolutionary framework, this allows us to link alleles of interest to a particular biological function – a strategy that is bolstered with good physiological, ecological, or fitness-related evidence to support it. In this way, we can better understand the intricate connections between genetic variation, phenotypic variation, and fitness to obtain a holistic view of the evolutionary processes affecting a species or population. But how can we apply this approach to conservation management?
Applications in conservation contexts
For the vast majority of conservation genomics studies, there is still a predominant focus on genome-wide patterns of genetic diversity and how this can guide conservation protocols – through defining population structure, assessing the genetic health of populations, or making recommendations for genetic rescue, for example. Compared to the long history of using neutral genetic markers to assess genetic diversity, the ability to link to specific genetic markers to adaptive or phenotypic traits is relatively recent. With the development of functional genomics tools, however, we are now more equipped than ever to make these links and apply this information to conservation research.
These approaches strengthen our ability to understand the adaptive potential of threatened species and expand on broad-scale patterns to improve conservation management. But how might we do that? Here are a few examples of how functional genomics can be applied to conservation management.
Genetic mechanisms of inbreeding depression
One notable issue in conservation genetics research is of course the looming threat of inbreeding depression – the low fitness consequences of a lack of genetic diversity within a population – in threatened species. Although low genetic diversity is broadly associated with prevalence in inbreeding depression, thresholds are difficult to predict and typically not consistent across taxa – it is hard to gauge whether a species is suffering (or will suffer) from inbreeding depression from genome-wide patterns of genetic diversity alone. Instead, inbreeding depression is likely more closely linked to specific functional loci with deleterious mutations: notable examples in threatened species include an unknown gene causing chondrodystrophy in the California condor, the AKAP4 gene leading to deformed spermatozoa in cheetah and five genes linked to wild Soay sheep lamb survival rates.
A related concept is that of genetic load, which describes the accumulation of deleterious mutations within populations as they decrease in size. These deleterious mutations typically occur within large populations at relatively low frequency – their impacts on fitness are masked by another less-detrimental allele in heterozygous individuals. When populations suddenly reduce in size, however, there is greater opportunity for individuals to become homozygous for the deleterious allele due to inbreeding, preventing the masking effect from happening and causing significant fitness effects.

An example of how genetic load accumulated in a bottlenecked population of fish, based on one locus with a dominant neutral/adaptive allele (green) and a recessive deleterious allele (red). In this scenario, being homozygous for the red allele would have major fitness impacts (say, fatality). The initial population retains the recessive allele at low frequency because its impact is masked by the dominant allele in the heterozygous individuals – there is no fitness cost associated with the allele in these individuals. However, when the population rapidly declines, inbreeding increases and causes greater homozygosity within the population. Fish which obtain two copies of the red allele now have major fitness consequences as a form of inbreeding depression. These dynamics may play out over many loci simultaneously, contributing to the overall “genetic load” of the population.
But there is some hope for small populations, with evidence that some might be able to ‘purge’ these deleterious alleles over time. Once exposed (as homozygotes) in the small population, natural selection can act more directly against the variant and, over time, it might become ‘bred out’ of the population. The efficacy of populations to be able to purge deleterious mutations likely dependent on the genetic basis of inbreeding depression, and thus better understanding which functional loci are responsible for inbreeding depression is important to predict the risk of genetic load and inbreeding depression in threatened populations and make management recommendations accordingly.

An example of how long-term small populations can purge genetic load. Because small (inbred) populations are more likely to possess the deleterious (red) allele in a homozygous state, which has major fitness consequences, the allele may (over time) be removed from the population. However, a few heterozygotes (possessing both red and green alleles) while mask this impact and it will be maintained in the population at low frequency (like the larger population above). Exactly how effectively purging can remove genetic load is somewhat debatable, but might explain how small populations can sometimes persist for a long time.
Mechanisms of adaptation
Conversely, functional genomics also allows us to better associate genetic variation with adaptive traits, identifying the genetic basis of important traits related to physiology or reproduction, for example. The association of adaptive loci to biological functions, phenotypic traits and ultimately measures of fitness allows us to better to understand the finer mechanisms of adaptive evolution in species.

An example of using functional genomics to understand a classic example of adaptive evolution – the peppered moth. During the industrial revolution, rare black morph (carbonaria) moths become more abundant relative to wild-type white moths due to changes in the selective environment. However, it wasn’t until van’t Hof et al. 2011 that the genomic region responsible for the melanism of black moths was discovered – a single region similar to silkworm chromosome 17. Several genes in this region have been shown to be responsible for colour patterns in another iconic lepidopteran group – Heliconius butterflies – suggesting that colour evolution in a wide range of species might have a surprisingly simple and shared basis.
The more thorough understanding of adaptation that functional genomics provides also leads to a greater understanding of the adaptive potential and resilience of populations, particularly those with low genetic diversity. As the impacts of climate change become more prevalent and obvious across the globe, functional genomics approaches can also help us understand how likely (indeed, if at all) populations will be able to adapt to rapidly changing environments. In terms of direct conservation management actions, this information will be critical in guiding genetic rescue and adaptive introgression across particularly threatened populations to promote resilience to these shifting conditions. For example, identifying adaptive diversity and how it relates to environmental variables might allow us to better predict how well an individual which is being translocated might match a target population – in this way, we can choose to translocate individuals which are most genetically adapted to the target conditions as a more targeted management action.
Disease genomics in threatened species
Species with critically low genetic diversity in functional genes (often caused by recent and strong genetic bottlenecks) may also be more susceptible to disease. High-profile (mammalian) examples include the transmissible Devil Facial Tumour Disease in Tasmanian devils; high levels of chlamydia infection in koalas; and broad susceptibility in Californian sea lions. Higher susceptibility to parasites and infectious disease in genetically depauperate species is often linked to a lack of genetic diversity within important immune function genes. For example, the major histocompatibility complex is a well-studied group of genes which influence several phenotypic traits, including (and especially) the immune response of organisms. A lack of MHC diversity is often linked to higher prevalence and susceptibility to disease, although this pattern is not always consistent or predictable.

The outcome of a transmissible cancer causing Devil Facial Tumour Disease. Image source: Menna Jones via Wikipedia (CC BY 2.5).
Conversely, individuals with higher genetic diversity (through lower internal relatedness and inbreeding) may provide greater resistance to infectious disease through innate immunity, a factor which likely played a deterministic role in the survival of some bottlenose dolphins to a morbillivirus outbreak. Thus, for species with critically low genetic diversity, introducing new genetic variation from other populations (through genetic rescue) may provide a key avenue to address disease susceptibility.
The future of functional approaches
Functional genomics is a growing discipline with expanding applications for conservation management. In the coming years, the intricacies of the example topics above will likely come further to light, with a better understanding of the functional basis of inbreeding depression, adaptive potential, and disease susceptibility in threatened populations. Of course, this is only a snapshot of some of the potential of the approach, and many others currently exist (or will soon exist) which may help improve our genomics-based approaches of species conservation.

One thought on “Conservation applications of functional variation”