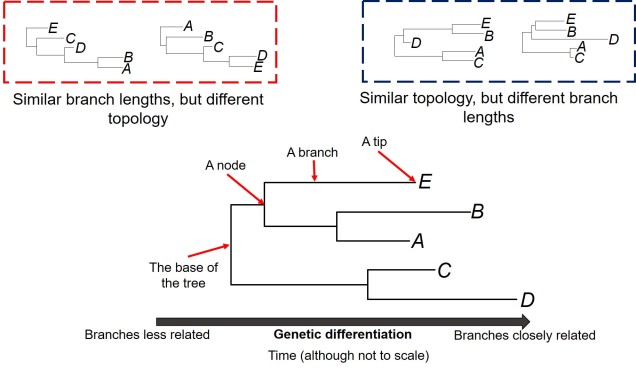
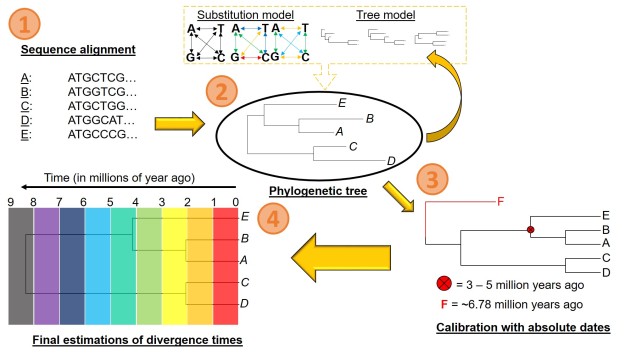
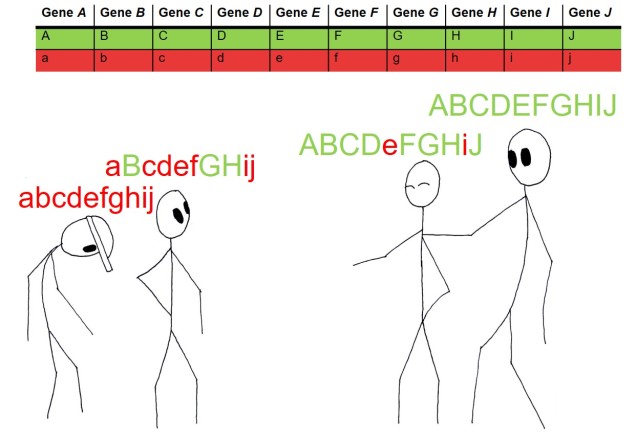
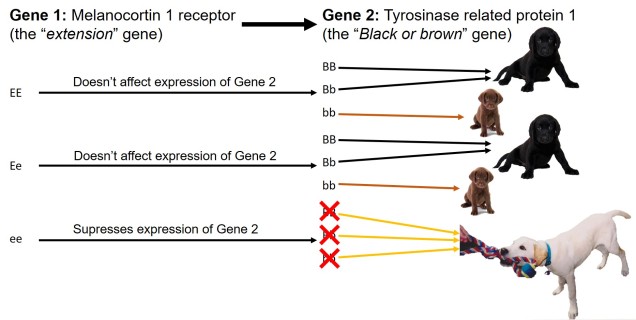
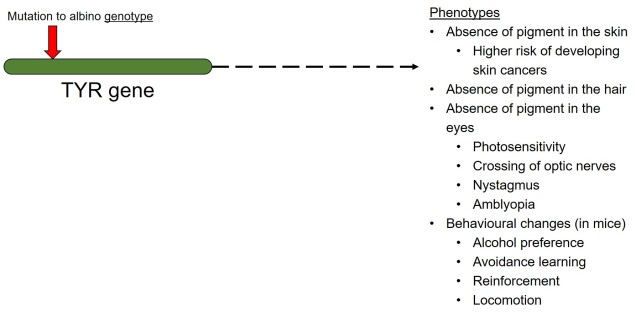
Biogeography of the globe
The distribution of organisms across the Earth, both over time and across space, is a fundamental aspect of the field of biogeography. But our understanding of the mechanisms by which organisms are distributed across the globe, and how this affects their evolution, can be at times highly enigmatic. Why are Australia and the Americas the only two places that have marsupials? How did lemurs get all the way to Madagascar, and why are they the only primate that has made the trip? How did Darwin’s famous finches get over to the Galápagos, and why are there so many species of them there now?
All of these questions can be addressed with a combination of genetic, environmental and ecological information across a variety of timescales. However, the overall field of biogeography (and phylogeography as a derivative of it) has traditionally been largely rooted on a strong yet changing theoretical basis. The earliest discussions and discoveries related to biogeography as a field of science date back to the 18th Century, and to Carl Linnaeus (to whom we owe our binomial classification system) and Alexander von Humboldt. These scientists (and undoubtedly many others of that era) were among the first to notice how organisms in similar climates (e.g. Australia, South Africa and South America) showed similar physical characteristics despite being so distantly separated (both in their groups and geographic distance). The communities of these regions also appeared to be highly similar. So how could this be possible over such huge distances?

Dispersal or vicariance?
Two main explanations for these patterns are possible; dispersal and vicariance. As one might expect, dispersal denotes that an ancestral species was distributed in one of these places (referred to as the ‘centre of origin’) before it migrated and inhabited the other places. Contrastingly, vicariance suggests that the ancestral species was distributed everywhere originally, covering all contemporary ranges within it. However, changes in geography, climate or the formation of other barriers caused the range of the ancestor to fragment, with each fragmented group evolving into its own distinct species (or group of species).
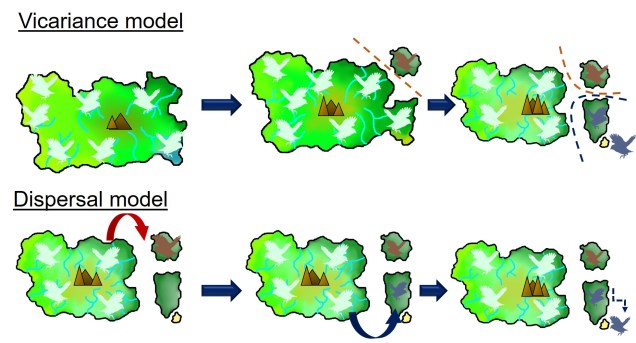
In initial biogeographic science, dispersal was the most heavily favoured explanation. At the time, there was no clear mechanism by which organisms could be present all over the globe without some form of dispersal: it was generally believed that the world was a static, unmoving system. Dispersal was well supported by some biological evidence such as the diversification of Darwin’s finches across the Galápagos archipelago. Thus, this concept was supported through the proposals of a number of prominent scientists such as Charles Darwin and A.R. Wallace. For others, however, the distance required for dispersal (such as across entire oceans) seemed implausible and biologically unrealistic.
A paradigm shift in biogeography
Two particular developments in theory are credited with a paradigm shift in the field; cladistics and plate tectonics. Cladistics simply involved using shared biological characteristics to reconstruct the evolutionary relationships of species (think like phylogenetics, but using physical traits instead of genetic sequence). Just as importantly, however, was plate tectonic theory, which provided a clear way for organisms to spread across the planet. By understanding that, deep in the past, all continents had been directly connected to one another provides a convenient explanation for how species groups spread. Instead of requiring for species to travel across entire oceans, continental drift meant that one widespread and ancient ancestor on the historic supercontinent (Pangaea; or subsequently Gondwana and Laurasia) could become fragmented. It only required that groups were very old, but not necessarily very dispersive.

From these advances in theory, cladistic vicariance biogeography was born. The field rapidly overtook dispersal as the most likely explanation for biogeographic patterns across the globe by not only providing a clear mechanism to explain these but also an analytical framework to test questions relating to these patterns. Further developments into the analytical backbone of cladistic vicariance allowed for more nuanced questions of biogeography to be asked, although still fundamentally ignored the role of potential dispersals in explaining species’ distributions.
Modern philosophy of biogeography
So, what is the current state of the field? Well, the more we research biogeographic patterns with better data (such as with genomics) the more we realise just how complicated the history of life on Earth can be. Complex modelling (such as Bayesian methods) allow us to more explicitly test the impact of Earth history events on our study species, and can provide more detailed overview of the evolutionary history of the species (such as by directly estimating times of divergence, amount of dispersal, extent of range shifts).
From a theoretical perspective, the consistency of patterns of groups is always in question and exactly what determines what species occurs where is still somewhat debatable. However, the greater number of types of data we can now include (such as geological, paleontological, climatic, hydrological, genetic…the list goes on!) allows us to paint a better picture of life on Earth. By combining information about what we know happened on Earth, with what we know has happened to species, we can start to make links between Earth history and species history to better understand how (or if) these events have shaped evolution.