The genetics of adaptation
Adaptation and evolution by natural selection remains one of the most significant research questions in many disciplines of biology, and this is undoubtedly true for molecular ecology. While traditional evolutionary studies have been based on the physiological aspects of organisms and how this relates to their evolution, such as how these traits improve their fitness, the genetic component of adaptation is still somewhat elusive for many species and traits.
Hunting for adaptive genes in the genome
We’ve previously looked at the two main categories of genetic variation: neutral and adaptive. Although we’ve focused predominantly on the neutral components of the genome, and the types of questions about demographic history, geographic influences and the effect of genetic drift, they cannot tell us (directly) about the process of adaptation and natural selective changes in species. To look at this area, we’d have to focus on adaptive variation instead; that is, genes (or other related genetic markers) which directly influence the ability of a species to adapt and evolve. These are directly under natural selection, either positively (‘selected for’) or negatively (‘selected against’).
Given how complex organisms, the environment and genomes can be, it can be difficult to determine exactly what is a real (i.e. strong) selective pressure, how this is influenced by the physical characteristics of the organism (the ‘phenotype’) and which genes are fundamental to the process (the ‘genotype’). Even determining the relevant genes can be difficult; how do we find the needle-like adaptive genes in a genomic haystack?

There’s a variety of different methods we can use to find adaptive genetic variation, each with particular drawbacks and strengths. Many of these are based on tests of the frequency of alleles, rather than on the exact genetic changes themselves; adaptation works more often by favouring one variant over another rather than completely removing the less-adaptive variant (this would be called ‘fixation’). So measuring the frequency of different alleles is a central component of many analyses.
FST outlier tests
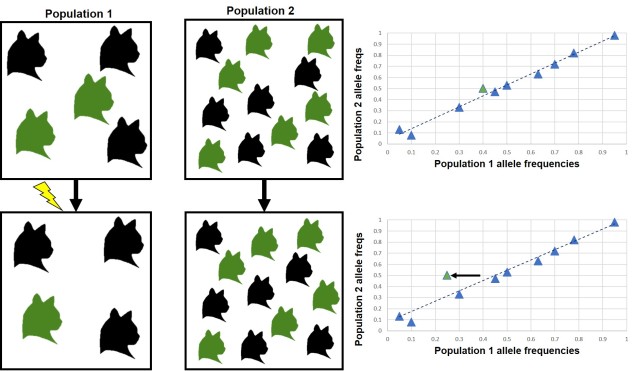
One of the most classical examples is called an ‘FST outlier test’. This can be a bit complicated without understanding what FST is actually measures: in short terms, it’s a statistical measure of ‘population differentiation due to genetic structure’. The FST value of one particular population can determine how genetically similar it is to another. An FST value of 1 implies that the two populations are as genetically different as they could possibly be, whilst an FST value of 0 implies that they are genetically identical populations.
Generally, FST reflects neutral genetic structure: it gives a background of how, on average, different are two populations. However, if we know what the average amount of genetic differentiation should be for a neutral DNA marker, then we would predict that adaptive markers are significantly different. This is because a gene under selection should be more directly pushed towards or away from one variant (allele) than another, and much more strongly than the neutral variation would predict. Thus, the alleles that are way more or less frequent than the average pattern we might assume are under selection. This is the basis of the FST outlier test; by comparing two or more populations (using FST), and looking at the distribution of allele frequencies, we can pick out a few alleles that vary from the average pattern and suggest that they are under selection (i.e. are adaptive).
There are a few significant drawbacks for FST outlier tests. One of the most major ones is that genetic drift can also produce a large number of outliers; in a small population, for example, one allele might be fixed (has a frequency of 1, with no alternative allele in the population) simply because there is not enough diversity or population size to sustain more alleles. Even if this particular allele was extremely detrimental, it’d still appear to be favoured by natural selection just because of drift.
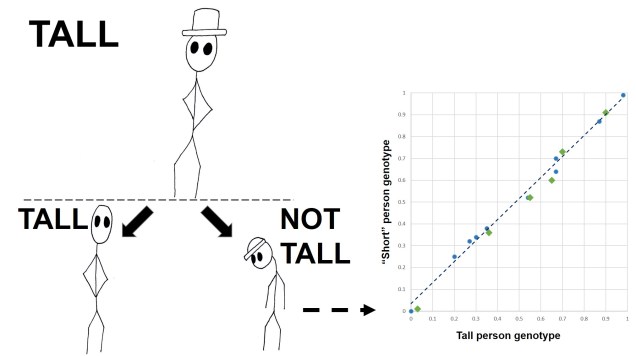
Secondly, the cut-off for a ‘significant’ vs. ‘relatively different but possibly not under selection’ can be a bit arbitrary; some genes that are under weak selection can go undetected. Furthermore, recent studies have shown a growing appreciation for polygenic adaptation, where tiny changes in allele frequencies of many different genes combine together to cause strong evolutionary changes. For example, despite the clear heritable nature of height (tall people often have tall children), there is no clear ‘height’ gene: instead, it appears that hundreds of genes are potentially very minor height contributors.
Genotype-environment associations
To overcome these biases, sometimes we might take a more methodological approach called ‘genotype-environment association’. This analysis differs in that we select what we think our selective pressures are: often environmental characteristics such as rainfall, temperature, habitat type or altitude. We then take two types of measures per individual organism: the genotype, through DNA sequencing, and the relevant environmental values for that organisms’ location. We repeat this over the full distribution of the species, taking a good number of samples per population and making sure we capture the full variation in the environment. Then we perform a correlation-type analysis, which seeks to see if there’s a connection or trend between any particular alleles and any environmental variables. The most relevant variables are often pulled out of the environmental dataset and focused on to reduce noise in the data.
The main benefit of GEA over FST outlier tests is that it’s unlikely to be as strongly influenced by genetic drift. Unless (coincidentally) populations are drifting at the same genes in the same pattern as the environment, the analysis is unlikely to falsely pick it up. However, it can still be confounded by neutral population structure; if one population randomly has a lot of unique alleles or variation, and also occurs in a somewhat unique environment, it can bias the correlation. Furthermore, GEA is limited by the accuracy and relevance of the environmental variables chosen; if we pick only a few, or miss the most important ones for the species, we won’t be able to detect a large number of very relevant (and likely very selective) genes. This is a universal problem in model-based approaches and not just limited to GEA analysis.
New spells to find adaptive genes?
It seems likely that with increasing datasets and better analytical platforms, many more types of analysis will be developed to delve deeper into the adaptive aspects of the genome. With whole-genome sequencing starting to become a reality for non-model species, better annotation of current genomes and a steadily increasing database of functional genes, the ability of researchers to investigate evolution and adaptation at the genomic level is also increasing.

10 thoughts on “Fantastic Genes and Where to Find Them”