The pygmy perches
I’ve mentioned a few times in the past that my own research centres around a particular group of fish: the pygmy perches. When I tell people about them, sometimes I get the question “why do you want to study them?” And to be fair, it’s a good question: there must be something inherently interesting about them to be worth researching. And there is plenty.
Pygmy perches are a group of very small (usually 4-6cm) freshwater fish native to temperate Australia: they’re found throughout the southwest corner of WA and the southeast of Australia, stretching from the mouth of the Murray River in SA up to lower Queensland (predominantly throughout the Murray-Darling Basin) and even in northern Tasmania. There’s a massive space in the middle where they aren’t found: this is the Nullarbor Plain, and is a significant barrier for nearly all freshwater species (since it holds practically no water).
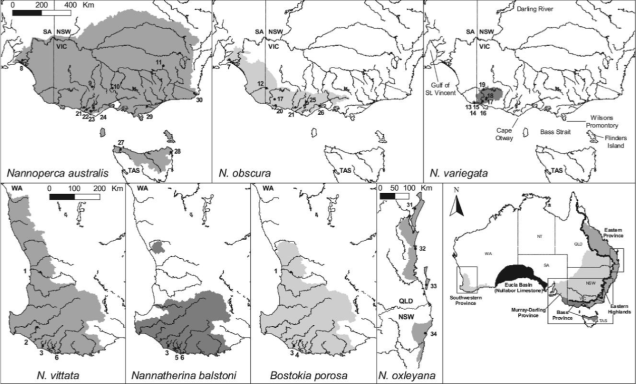
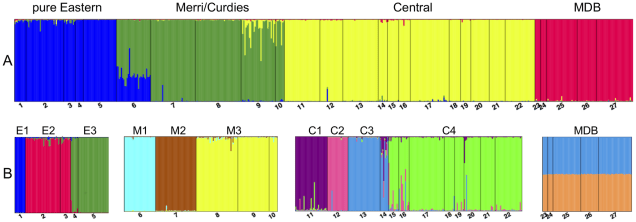
The group consists of 2 genera (Nannoperca and Nannatherina) and 7 currently described species, although there could be as many as 10 actual species (see ‘cryptic species’: I’ll elaborate on this more in future posts…). They’re very picky about their habitat, preferring to stay within low flow waterbodies with high vegetation cover, such as floodplains and lowland creeks. Most species have a lifespan of a couple years, with different breeding times depending on the species.
Why study pygmy perches?
So, they’re pretty cute little fish. But unfortunately, that’s not usually enough justification to study a particular organism. So, why does the Molecular Ecology Lab choose to use pygmy perch as one (of several) focal groups? Well, there’s a number of different reasons.
The main factors that contribute to their research interest are their other characteristics: because they’re so small and habitat specialists, they often form small, isolated populations that are naturally separated by higher flow rivers and environmental barriers. They also appear to have naturally very low genetic diversity: ordinarily, we’d expect that they wouldn’t be great at adapting and surviving over a long time. Yet, they’ve been here for a long time: so how do they do it? That’s the origin of many of the research questions for pygmy perches.
Adaptive evolution despite low genetic variation
One of the fundamental aspects of the genetic basis of evolution is the connection between genetic diversity and ‘adaptability’: we expect that populations or species with more genetic diversity are much more likely to be able to evolve and adapt to new selective pressures than those without it. Pygmy perches clearly contradict this at least a little bit, and so much of the research in the lab is about understanding exactly what factors and mechanisms contribute to the ability of pygmy perches to apparently adapt and survive what is traditionally not consider a very tolerant place to live. Recent research suggests the different expression of genes may be an important mechanism of adaptation for pygmy perch.
Recommended readings: Brauer et al. (2016); Brauer et al. (2017).
The influence of the historic environment on evolution
From an evolutionary standpoint, pygmy perches are unique in more ways than just their genetic diversity. They’re relatively ancient, with the origin of the group estimated at around 40 million years ago. Since then, they’ve diversified into a number of different species and have spread all over the southern half of the Australian continent, demonstrating multiple movements across Australia in that time. This pattern is unusual for freshwater organisms, and this combined with their ancient nature makes them ideal candidates for studying the influence of historic environment, climate and geology on the evolution and speciation of freshwater animals in Australia. And that’s the focus of my PhD (although not exclusively; plenty of other projects have explored questions in this area).
Recommended readings: Unmack et al. (2013); Unmack et al. (2011).
Conservation management and ecological role
Of course, it’s all well and good to study the natural, evolutionary history of an organism as if it hasn’t had any other influences. But we all know how dramatic the impact humans have on the environment are and unfortunately for many pygmy perch species this means that they are threatened or endangered and at risk of extinction. Their biggest threats are introduced predators (such as the redfin perch and European carp), alteration of waterways (predominantly for agriculture) and of course, climate change. For some populations, local extinction has already happened: some populations of the Yarra pygmy perch (N. obscura) are now completely gone from the wild. Many of these declines occurred during the Millennium Drought, where the aforementioned factors were exacerbated by extremely low water availability and consistently high temperatures. So naturally, a significant proportion of the work on pygmy perches is focused on their conservation, and trying to boost and recover declining populations.
This includes the formation of genetics-based breeding programs for two species, the southern pygmy perch and Yarra pygmy perch. A number of different organisations are involved in this ongoing process, including a couple of schools! These programs are informed by our other studies of pygmy perch evolution and adaptive potential and hopefully combined we can save these species from becoming totally extinct.


Recommended readings: Brauer et al. (2013); Attard et al. (2016); Hammer et al. (2013).
Hopefully, some of this convinces you that pygmy perch are actually rather interesting creatures (I certainly think so!). Pygmy perch research can offer a unique insight into evolutionary history, historical biogeography, and conservation management. Also, they’re kinda cute….so that’s gotta count for something, right? If you wanted to find out more about pygmy perch research, and get updates on our findings, be sure to check out the Molecular Ecology Lab Facebook page or our website!