What is a ‘molecular marker’?
As we’ve previously discussed within The G-CAT, information from the DNA of organisms can be used in a variety of ways to study evolution and ecology, inform conservation management, and understand the diversity of life on Earth. We’ve also had a look at the general background of the DNA itself, and some of the different parts of the genome. What we haven’t discussed yet is how we use the DNA sequence in these studies; most importantly, which part of the genome to use.
The genome of most organisms is massive. The size of the genome ranges depending on the organism, with one of the smallest recorded genomes belonging to a bacteria (Carsonella ruddi), consisting of 160,000 bases. There is a bit of debate about the largest recorded genome, but one contender (the ‘canopy plant’, Paris japonica) has a genome stretching 150 billion base pairs long! The human genome sits in the middle at around 3 billion bases long. Naturally, it would be incredibly difficult to obtain the sequence of the whole genome of many organisms (particularly 20 – 30 years ago, due to technological limitations in the sequencing process) so instead we usually pick a specific region of the genome instead. The exact region (or type of region) we use is referred to as a ‘molecular marker’.
How do we choose a good marker?
The marker we pick is incredibly important: this is often based on how much variation we need to observe across groups. For example, if we want to study differences between individuals, say in a pedigree analysis, we need to pick a section of the DNA that will show differences between individuals; it will need to mutate fairly rapidly to be useful. If it mutates too slowly, all individuals will look identical genetically and we won’t have learnt anything new at all.
On the flipside, if we want to study evolution at a larger scale (say, between species, or groups of species) we would need to use a marker that evolves much slower. Using a rapidly mutating section of DNA would effectively give a tonne of ‘white noise’; it’d be impossible to pick what is the genetic difference at the species level (i.e. one species is different to another at that base) vs. at the individual level (i.e. one or many individuals within the species are different). Thus, we tend to use much slower mutating markers for deeper evolutionary history.

Think of it like comparing cats and dogs. If we wanted to compare different cats to one another (say different breeds) we could use hair length or coat colour as a useful trait. Since some breeds have different coat characteristics, and these don’t vary as much within the breed as across breeds, we can easily determine a long haired cat from a short haired cat. However, if we tried to use coat colour and length to compare cats and dogs we’d be stumped, because both species have lots of variation in these traits within their species. Some cats have coat length more similar to some dogs than to other cats for example; so they’re not a good characteristics to separate the two animal species (we might use muzzle shape, or body shape instead). If we substitute each of these traits with a particular marker, then we can see that some markers are better for some comparisons but not good for others.
Allozymes
The most traditional molecular marker are referred to as ‘allozymes’; instead of comparing actual genetic sequences (something that was not readily possible early in the field), variations in the shape (i.e. the amino acids of the protein, not the code underlying it) were compared between species. Changes in proteins occur very rarely as natural selection tends to push against randomly changing protein structure, since the shape of it is critical to its function and functionality. Because of this, allozymes were only really effective for studying very broad comparisons (mainly across species or species groups); the exact protein used depends on the study organism. Allozymes are generally considered outdated in the field nowadays.
With the development of technologies that allowed us to actual determine the DNA code of genes, molecular ecology moved into comparing actual sequences across individuals. However, early sequencing technology could generally only accurately determine small sections of DNA at a time, so particular markers capitalising on this were developed. Many of these are still used due to their cost-effectiveness and general ease of analysing.
Microsatellites
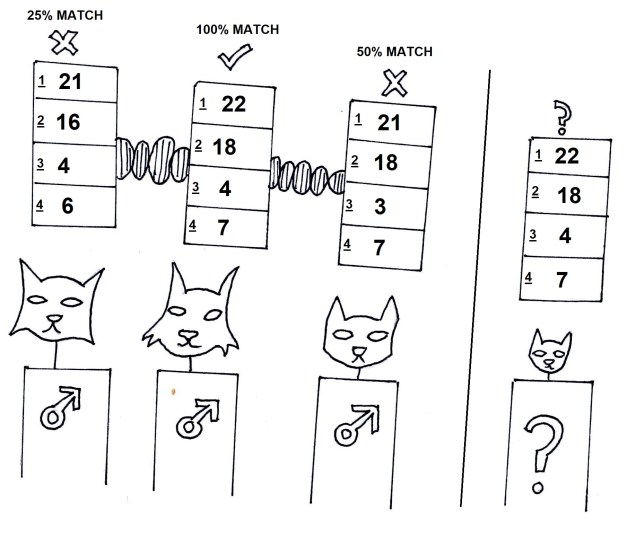
For comparing closely related individuals (within a pedigree, or a population), markers called ‘microsatellites’ are widely used. These are small sections of the genome which have repetitive DNA codes; usually, the same two or three base pairs (one ‘motif’) are repeated a number of times afterwards (the ‘repeat number’). While the motifs themselves rarely get mutations, the number of repeated motifs very rapidly mutates. This is because the protein that copies DNA is not very perfect, and often ‘slips up’, and adds or cuts off a repeat from the microsatellite sequence. Thus, differences in the repeat number of microsatellites accumulate pretty quickly, to the point where you can determine the parents of an individual with them.
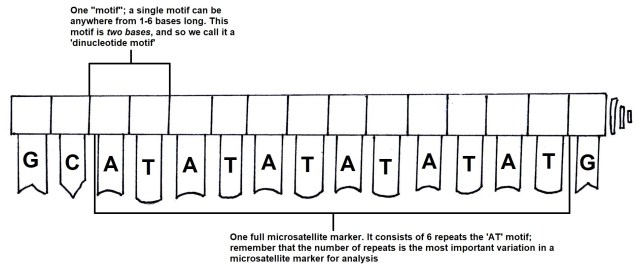
Microsatellites are often used in comparisons across closely related individuals, such as within pedigrees or within populations. While they are relatively easy to obtain, one drawback is that you need to have some understanding of the exact microsatellite you wish to analyse before you start; you need to make a specific ‘primer’ sequence to be able to get the right marker, as some may not be informative in particular species or comparisons. Many researchers choose to use 10-20 different microsatellite markers together in these types of studies, such as in human parentage analyses.
Mitochondrial DNA
For deeper comparisons, however, microsatellites mutate far too rapidly to be effective. Instead, we can choose to use the DNA of the mitochondria. You may remember the mitochondria as ‘the powerhouse of the cell’; while this is true, it also has a lot of other unique properties. The mitochondria was actually (a very, very, very long time ago) a separate bacteria-like organism which became symbiotically embedded within another cell. Because of this, and despite a couple billion years of evolution since that time, the mitochondria actually has its own genome separate to the ‘host’ (like the standard human genome). The full mitochondrial genome consists of around 37 different genes, most of which don’t code for any proteins involved directly in evolution; as such, natural selection doesn’t affect them as much as other genes. The most commonly used mitochondrial genes are the cytochrome b oxidase gene (cytb for short) or the cytochrome c oxidase 1 (CO1) gene.
The mitochondrial genome evolves relatively rapidly (but not nearly as fast as microsatellites) and is found in pretty much every plant and animal on the planet. Because of these traits, it’s often used as a way of diagnosing species through the ‘Barcode of Life’ project (using cytb and CO1). It’s very widely used within species-level studies, to the point where we can even use the relatively consistent mutation rate of the mitochondrial genome to estimate how long ago different species separated in evolution.

Other markers?
There are plenty of other genetic markers that are used within molecular ecology, with some focusing on only the exons or introns of genes, or other repetitive sequences. However, microsatellites and mitochondrial genes are among the most widely used in evolution and conservation studies.
While these markers have been very useful in building the foundations of molecular ecology as a scientific field, developments in sequencing technology, analytical methods and evolutionary theory have pushed our ability to use DNA to understand evolution and conservation even further. Particularly the development of sequencing machines which can process much larger amounts of genetic DNA. This has pushed genetics into the age of ‘genomics’: while this sounds like a massively technical difference, it’s really just about the difference in the size of the data we can use. Obviously, this has many other benefits for the kinds of questions we can ask about evolution, conservation and ecology.
Genomics has massively expanded in recent years, the types, quantity and quality of data are diverse. Stay tuned because next week, we’ll start to delve into the modern world of genomics!
