Evolution travelling in time
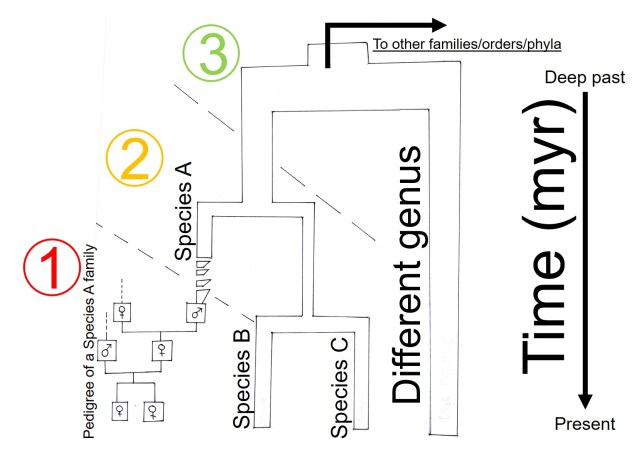
As I’ve mentioned a few times before, evolution is a constant force that changes and flows over time. While sometimes it’s more convenient to think of evolution as a series of rather discrete events (a species pops up here, a population separates here, etc.), it’s really a more continual process. The context and strength of evolutionary forces, such as natural selection, changes as species and the environment they inhabit also changes. This is important to remember in evolutionary studies because although we might think of more recent and immediate causes of the evolutionary changes we see, they might actually reflect much more historic patterns. For example, extremely low contemporary levels of genetic diversity in cheetah is likely largely due to a severe reduction in their numbers during the last ice age, ~12 thousand years ago (that’s not to say that modern human issues haven’t also been seriously detrimental to them). Similarly, we can see how the low genetic diversity of a small population colonise a new area can have long term effects on their genetic variation: this is called ‘founder effect’. Because of this, we often have to consider the temporal aspect of a species’ evolution.

Evolution travelling across space
If the environmental context of species and populations are also important for determining the evolutionary pathways of organisms, then we must also consider the spatial context. Because of this, we also need to look at where evolution is happening in the world; what kinds of geographic, climatic, hydrological or geological patterns are shaping and influencing the evolution of species? These patterns can influence both neutral or adaptive processes by shaping exactly how populations or species exist in nature; how connected they are, how many populations they can sustain, how large those populations can sustainably become, and what kinds of selective pressures those populations are under.
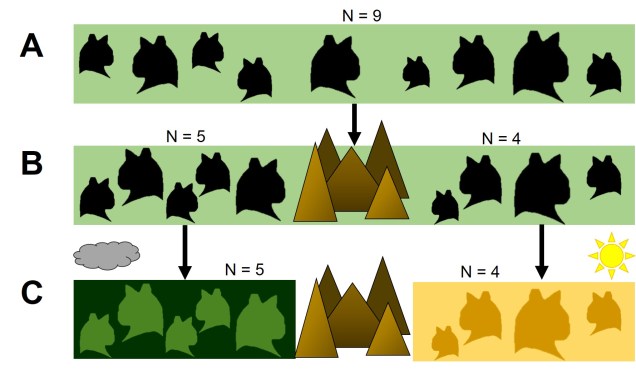
Evolution along the space-time continuum
Given that the environment also changes over time (and can be very rapid, and we’ve seen recently), the interaction of the spatial and temporal aspects of evolution are critical in understanding the true evolutionary history of species. As we know, the selective environment is what determines what is, and isn’t, adaptive (or maladaptive), so we can easily imagine how a change in the environment could push changes in species. Even from a neutral perspective, geography is important to consider since it can directly determine which populations are or aren’t connected, how many populations there are in total or how big populations can sustainably get. It’s always important to consider how evolution travels along the space-time continuum.

Phylogeography
The field of evolutionary science most concerned with these two factors and how the influence evolution is known as ‘phylogeography’, which I’ve briefly mentioned in previous posts. In essence, phylogeographers are interested in how the general environment (e.g. geology, hydrology, climate, etc) have influenced the distribution of genealogical lineages. That’s a bit of a mouthful and seems a bit complicated, by the genealogical part is important; phylogeography has a keen basis in evolutionary genetics theory and analysis, and explicitly uses genetic data to test patterns of historic evolution. Simply testing the association between broad species or populations, without the genetic background, and their environment, falls under the umbrella field of ‘biogeography’. Semantics, but important.
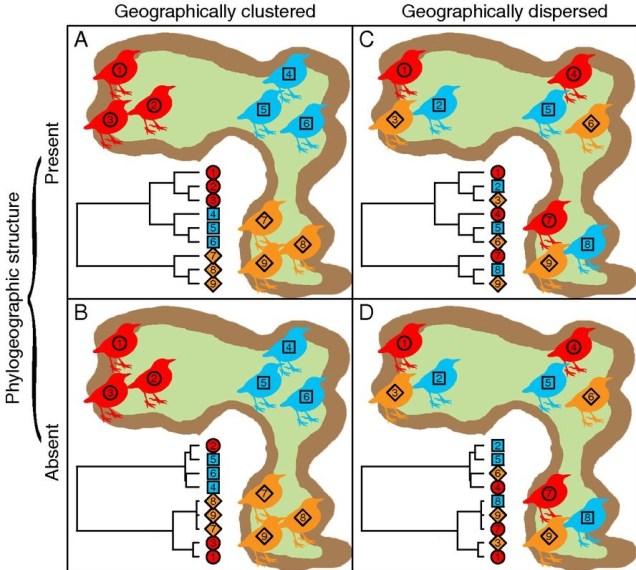
For phylogeography, the genetic history of populations or species gives the more accurate overview of their history; it allows us to test when populations or species became separated, which were most closely related, and whether patterns are similar or different across other taxonomic groups. Predominantly, phylogeography is based on neutral genetic variation, as using adaptive variation can confound the patterns we are testing. Additionally, since neutral variation changes over time in a generally predictable, mathematical format (see this post to see what I mean), we can make testable models of various phylogeographic patterns and see how well our genetic data makes sense under each model. For example, we could make a couple different models of how many historic populations there were and see which one makes the most sense for our data (with a statistical basis, of course). This wouldn’t work with genes under selection since they (by their nature) wouldn’t fit a standard ‘neutral’ model.

That said, there are plenty of interesting scientific questions within phylogeography that look at exploring the adaptive variation of historic populations or species and how this has influenced their evolution. Although this can’t inherently be built into the same models as the neutral patterns, looking at candidate genes that we think are important for evolution and seeing how their distributions and patterns relate to the overall phylogeographic history of the species is one way of investigating historic adaptive evolution. For example, we might track changes in adaptive genes by seeing which populations have which variants of the gene and referring to our phylogeographic history to see how and when these variants arose. This can help us understand how phylogeographic patterns have influenced the adaptive evolution of different populations or species, or inversely, how adaptive traits might have influenced the geographic distribution of species or populations.
Where did you come from and where will you go?
Phylogeographic studies can tell us a lot about the history of a species, and particularly how that relates to the history of the Earth. All organisms share an intimate relationship with their environment, both over time and space, and keeping this in mind is key for understanding the true evolutionary history of life on Earth.