Understanding genetic determinants
You’ve probably been exposed to one news headline or another in the recent past (let’s say the last 5 years) that reads something like “SCIENTISTS DISCOVER GENES THAT CAUSE (X).” X, of course, varies massively based on the study itself (and sometimes the bastardisation of said study by media): it can include describing medical conditions such as cancer, autism or congenital diseases; behavioural traits, such as sexual preferences; or broad physical traits, such as the classic problem of the inheritability of height. Unsurprisingly, you may think that trying to find the genes responsible for some traits should be either a) super easy, or b) super hard, depending on your own philosophical preference or the trait in question. So how do these studies come about, anyway?

Genome-wide association studies
All of these studies have one unifying framework underlying them: the notion of correlating certain alleles with a particular trait. Given the accessibility of whole genomes for humans broadly, these methods are rooted in the idea of scanning the full genome of people and trying to figure out which variations in the genome correlate best with the trait in question. These types of studies are naturally referred to as “genome-wide association studies”, or GWAS for short. They take a few different forms, and apply different statistical methods for inferring associations, but the basic principle is the same. These types of association studies are born of a particular empirical problem: that sometimes phenotypic (whether that be physical, psychological or ecological) traits which should be encoded by a particular gene show no obvious direct causative gene.
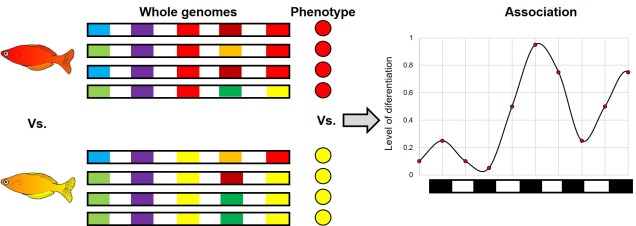
A classic example of this is in height. Although we know height clearly has a heritable component, it is difficult to determine a particular gene responsible. That is, there is no clear allele that, when possessed, leads to the development of taller humans regardless of the rest of their genetic background. Instead, we describe height as a polygenic trait: that is, very small changes across hundreds or thousands of different genes all act in combination to produce the ‘height’ trait of a person. Because height is a scale, and not a category, it is also sometimes referred to as being driven by ‘quantitative trait loci’, or QTLs. Because each individual allele may only very weakly contribute to the overall phenotypic response, determining causative genes from a background of natural genetic variation (noise) can be extremely difficult. This is the basis of performing genome-wide scans that use large datasets (for humans, often hundreds or thousands of people) to try and find correlations between the swathe of genetic variation and the trait in question.
The prevalence of GWAS seems to grow exponentially in time, predominantly driven by the increase availability of both genomic data (sometimes somewhat surreptitiously sourced from private enterprises like 23AndMe or Ancestry.com) and physiological/psychological data from surveys or medical reviews. While the root of GWAS is in understanding complex physiological traits such as height or development of diseases, where there is clear heritability but not obvious responsible gene, GWAS has since extended into other realms of inference. Here, we’ll take a look at some of these areas and their relevance to scientific enquiry.
Applications of GWAS
Medicine
Unsurprisingly, much of the drive and basis for GWAS is in medical research. Particularly, GWAS results are being used to try to determine genetic factors which may contribute or exacerbate the development of certain diseases. While this seems obvious for congenital diseases with a clear heritable component, GWAS are also applied to more complex scenarios such as the development of different types of cancer, trying to predict responses to different drug treatments, or identifying the risk of late-life degenerative diseases such as Parkinson’s and heart disease.

Psychology and behaviour
GWAS has also extended beyond medical applications, however. Particularly, there is a growing field of research dedicated to trying to understand genetic factors that may underlie certain psychological or behavioural traits in humans. This has a fairly broad spectrum – from understanding learning ability, to the basis of sexuality, and even to the interspecies communication ability of dogs to humans! – which will undoubtedly grow over time. Although there is some debate about the relative strength of genetic architecture in driving personality and psychological traits, the potential role of GWAS to try and address questions of the predictability and heritability of these traits is rapidly becoming a topic of interest.
The caveats of GWAS
Despite all of these potential applications, you might be surprised to note that there are some resounding concerns and disparaging opinions of GWAS, which incorporate both philosophical and empirical issues with the method.
Analytical caveats
As with most statistically-driven investigations (i.e. science), the power to rigorously detect a significant effect is a particular issue. With GWAS, a couple of different factors can drive issues with power and resolution in the study. Firstly, often massive amounts of data (mostly in terms of the number of samples – people – used in the study to obtain genome and phenotype data) are required to generate a statistically valuable result. This is because individual loci likely have weak associations with the trait in question, meaning that at smaller sample sizes they ‘blend in’ with the natural mosaic of background genetic variation.
Another similar issue based on the notion of weak correlations is the ‘causative power’ of the genes that appear significant. For many traits, the presence or absence of alleles (genetic variation) may explain very little of the variation in the trait, as a biological basis. The complex influence of environmental factors, and their interactions with the genetic architecture of a person, may account for a much greater proportion of the variation in the trait. In the case of height, developmental factors such as diet, exercise or other unknown external forces may obscure the impact of the underlying genetic factors.
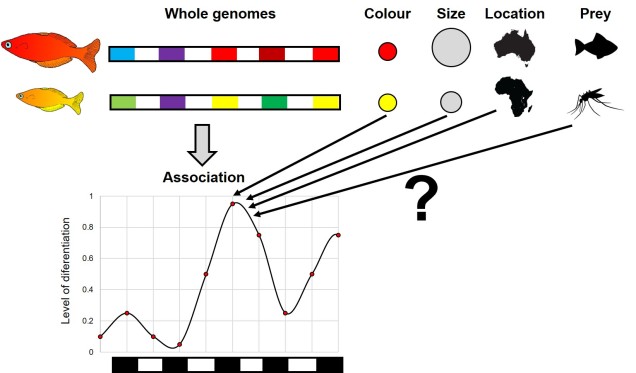
Another methodological concern with GWAS is the impact of correlations across various factors. Particularly, this often relates to the notion that allele frequencies across closely associated (‘linked’) genes will vary in tandem due to the impact of recombination. That is, for a particular gene acted upon by selection, neighbouring genes will also ‘appear’ to be experiencing selection, but are actually just copping the peripheral impact of the process. This aspect – referred to as linkage disequilibrium – was discussed in more detail here. In regards to GWAS, this might mean that we infer a particular gene as being ‘causative’, when it is actually just correlated with a gene responsible for our trait (indeed, if there is one).
A similar issue with correlation is based on the notion that the traits we wish to understand – physiological, psychological or otherwise – are also not always clearly defined and may correlate with other traits. A great work-through example of this is outlined in Graham Coop’s blog, using ‘tea-drinking’ as an example trait. Although I think he explains it better than I could, in essence this example demonstrates that comparing two populations (in this case French and English) for a single trait (in this case tea-drinking) means you might actually find genes that encode both tea-drinking and nationality. This gets more complicated if you consider the interactive effects (e.g. how allele frequencies vary over time in response to both the trait they might encode and the environment in which they occur). Pinpointing whether observed differences between the two populations are the result of beverage preferences or simply demographic history is a tricky process.
Philosophical concerns
Criticisms of the use of GWAS and polygenic scores extends beyond the empirical and enters the philosophical as well. Indeed, the notion that GWAS could be used to identify genetic variants responsible for certain characteristics, which could then be used to ‘screen’ people, is potentially concerning. Although this concern is multifaceted and largely involves the application of the results of GWAS, the notion of how information may be used unethically is not a new concept. For example, one concern is the potential for insurance companies to hike premiums for those deemed ‘at risk’ of developing various potentially preventable diseases. Another concern is that misinterpreted GWAS data (particularly in regards to polygenic risk scores from medical studies) may dramatically influence people’s behaviour in response.
Another equally relevant but even more ominous thought is the potential for prenatal screening of particular personality traits (including but not limited to homosexuality) that may open the door for eugenics practices. I consider Jeremy Yoder’s piece in Slate as essential reading in this regard: it encapsulates the complex nature of the ethics of these types of questions (and indeed, whether these questions are as informative as we expect).
To say that this is an ethical dilemma, one which cannot be ignored by scientific bodies responsible for the provision of this information, is an understatement.
The future of GWAS
Genome-wide association studies are a powerful tool to investigate the genetic basis behind many human (and non-human) traits, growing ever-more abundant and powerful as data and technology develop. However, as with all science, sensationalist claims and non-academic manipulation of a forefront field must be tempered by careful foresight and planning. What GWAS can do, can’t do, and should (or should do) is a complex array of issues that we must continue to evaluate.
