Sometimes when I talk about the concept of conservation genetics to friends and family outside of the field, there can be some confusion about what this actually means. Usually, it’s assumed that means the conservation of genetics: that is, instead of trying to conserve individual animals or plants, we try to conserve specific genes. While in some cases this is partially true (there might be genes of particular interest that we want to maintain in a wild population), often what we actually mean is using genetic information to inform conservation management and to give us the best chance of long-term rescue for endangered species.

See, the DNA of individuals contains much more information than just the genes that make up an organism. By looking at the number, frequency or distribution of changes and differences in DNA across individuals, populations or species, we can see a variety of different patterns. Typically, genetics-based conservation analysis is based on a single unifying concept: that different forces create different patterns in the genetic make-up of species and populations, and that these can be statistically evaluated using genetic data. The exact type or scale of effect depends on how the data is collected and what analysis we use to evaluate that data, although we could do multiple types of analysis using the same dataset.
Oftentimes, we want to know about the current or historical state of a species or population to best understand how to move forward: by understanding where a species has come from, what it has been affected by, and how it has responded to different pressures, we can start to suggest and best manage these species into the future.
However, there are lots of possible avenues for exploration: here are just a few…
Evolutionary significant units (ESUs) and management units (MUs)
One commonly used application of genetic information for conservation is the designation of what we call ‘Evolutionary Significant Units’ (ESUs). Using genetics, we can determine the boundaries of particular populations which correspond to their own unique evolutionary groups. These are often the results of historical processes which have separated and driven the independent evolution of each ESU, usually with low or no gene flow across these units. Generally, managing and conserving each of these can lead to overall more robust management of the species as a whole by making sure certain groups that have unique and potentially critical adaptations are maintained in the wild. Although ESUs can sometimes be arguable (particularly when there is some, but not much, gene flow across units), it forms an important aspect of conservation designations.
In cases of shorter term separations across these populations, where there are noticeable differences in the genetics of the populations but not necessarily massively different evolutionary histories, conservationists will sometimes refer to ‘Management Units’ (MUs). These have much weaker evolutionary pressure behind them but might be indicative of very recent impacts, such as human-driven fragmentation of habitat or contemporary climate change. MUs often reflect very sudden and recent changes in populations and might have profound implications for the future of these groups: thus, they are an important way of assessing the current state of the species. The next couple of figures demonstrate this from one of my colleagues’ research papers.

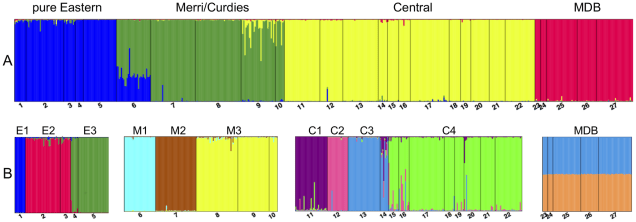
The two can be thought of as part of the same hierarchy, with ESUs reflecting more historic, evolutionary groups and MUs reflecting more recent (but not necessarily evolutionary) groups. For conservation management, this has traditionally meant that individuals from one ESU were managed independent of one another (to preserve their ‘pure’ evolutionary history) whilst translocations of individuals across MUs were common and often recommended. This is based on the idea that mixing very genetically different populations could cause adaptive genes in each population to become ‘diluted’, negatively affecting the ability of the populations to evolve: this is referred to as ‘outbreeding depression’ (OD).

However, more recent research has suggested that the concerns with OD from mixing across ESUs are less problematic than previously thought. Analysis of the effect of OD versus not supplementing populations with more genetic diversity has shown that OD is not the more dangerous option, and there is a current paradigm push to acknowledge the importance of mixing ESUs where needed.
Adaptive potential and future evolution
Understanding the genetic basis of evolution also forms an important research area for conservation management. This is particularly relevant for ‘adaptive potential’: that is, the ability for a particular species or population to be able to adapt to a variety of future stressors based on their current state. It is generally understood that having lots of different variants (alleles) of genes in the total population or species is a critical part of evolution: the more variants there are, the more choices there are for natural selection to act upon.
We can estimate this from the amount of genetic diversity within the population, as well as by trying to understand their previous experiences with adaptation and evolution. For example, it is predicted that species which occur in much more climatically variable habitats (such as in desert regions) are more likely to be able to handle and tolerate future climate change scenarios since they’ve demonstrated the ability to adapt to new, more extreme environments before. Examples of this include the Australian rainbowfishes, which are found in pretty well every climatic region across the continent (and therefore must be very good at adapting to new, varying habitats!).

Genetics-based breeding programs and pedigrees
A much more direct use of genetic information for conservation is in designing breeding programs. We know that breeding related individuals can have very bad results for offspring (this is referred to as ‘inbreeding depression’): so obviously, we would avoid breeding siblings together. However, in complex breeding systems (such as polygamous animals), or in wild populations, it can be very difficult to evaluate relationships and overall relatedness.
That’s where genetics comes in: by looking at how similar or different the DNA of two individuals are, we can not only check what relationship they are (e.g. siblings, cousins, or very distantly related) but also get an exact value of their genetic relatedness. Since we know that having a diverse gene pool is critical for future adaptation and survival of a species, genetics-based breeding programs can maximise the amount of genetic diversity in following generations. We can even use a computer algorithm to make the very best of breeding groups, using a quirky program called SWINGER.

Taxonomy for conservation legislation
Another (slightly more complicated) application of genetics is the designation of species status. Large amounts of genetic information can often clarify complex issues of species descriptions (later issues of The G-CAT will discuss exactly how this works and why it’s not so straightforward…).
Why should we care what we call a species or not? Well, much of the protective legislation at the government level is designed at the species-level: legislative protections are often designated for a particular species, but doesn’t often distinguish particular populations. Thus, misidentified species can sometimes but lost if they were never detected as a unique species (and assumed to be just a population of another species). Alternatively, managing two species as one based on misidentification could mess with the evolutionary pathways of both by creating unfit hybrid species which do not naturally come into contact together (say, breeding individuals from one species with another because we thought they were the same species).

Additionally, if we assume that multiple different species are actually only one species, this can provide an overestimate of how well that species is doing. Although in total it might look like there are plenty of individuals of the species around, if this was actually made of 4 separate species then each one would be doing ¼ as well as we thought. This can feed back into endangered status classification and thus conservation management.
These are just some of the most common examples of applied genetics in conservation management. No doubt going into the future more innovative and creative methods of applying genetic information to maintaining threatened species and populations will become apparent. It’s an exciting time to be in the field and inspires hope that we may be able to save species before they disappear from the planet permanently.



